Amyotrophic Lateral Sclerosis (ALS) Modeling & Pharmacodynamics Service
Amyotrophic lateral sclerosis is a neurodegenerative disorder leading to loss of voluntary muscle movement, paralysis, and eventually death. There is now an urgent need for the development of effective treatment for ALS. Creative Biolabs offers contract efficacy studies using standard transgenic SOD1 mice for the evaluation of putative therapeutics of ALS.
Introduction of ALS
Amyotrophic Lateral Sclerosis (ALS, or “Lou Gehrig's Disease”) is a progressive neurodegenerative disorder characterized by the degeneration of upper and lower motor neurons. Loss of these neurons leads to a deterioration of neuromuscular function, causing weakness, atrophy, and paralysis of skeletal muscles. The progression of ALS is usually rapid and death typically occurs within 3–5 years of diagnosis. Currently, there are no therapeutics that halt, delay or reverse the disease with the exception of riluzole which has a reproducibly modest effect on slowing disease progression in humans.
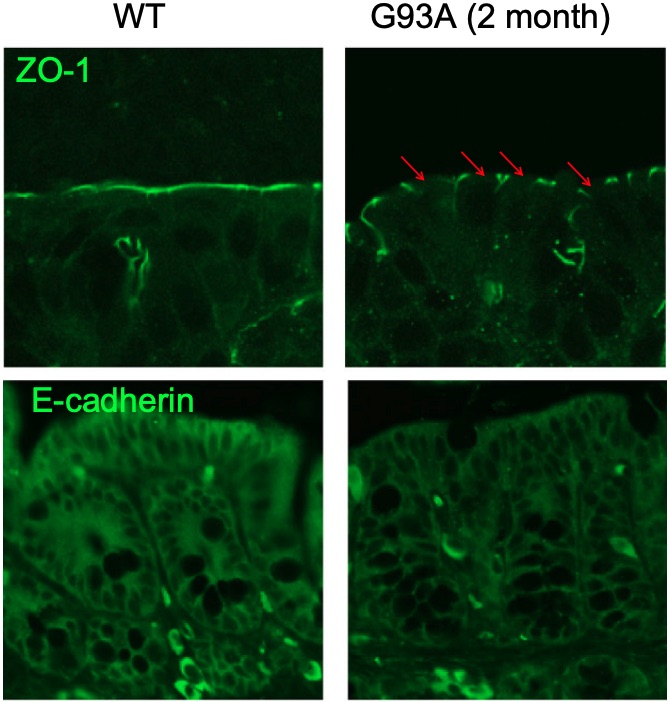 Fig. 1 Immunostaining of ZO-1 and E-cadherin in the colon of ALS model mice. 1
Fig. 1 Immunostaining of ZO-1 and E-cadherin in the colon of ALS model mice. 1
ALS can be familial (FALS, 10%), where the disease is propagated through the family, or sporadic (SALS, 90%), where no familial history of disease exists. The first gene shown to be mutated in FALS encodes the enzyme superoxide dismutase 1 (SOD1). Thereafter, mutations in other genes have also been found to be causative for the disease, such as repeat expansions in the gene coding C9ORF72. The discovery of these ALS causing genes has led to the development of primate, rodent, zebrafish, worm and fly ALS models which more or less mimic many, but not all aspects of disease seen in ALS patients. They are widely used for advancing our understanding of the pathophysiology of ALS as well as screening tools for testing molecules with therapeutic potential.
Features of SOD1-G93A Transgenic Mouse Model
- The transgenic mouse model (SOD1-G93A) of SOD1-ALS expresses approximately 20–24 copies of the human coding sequence with the G93A mutation, under control of the human SOD1 promoter.
- The mutation does not result in loss of activity of the enzyme, but rather the expression of a functioning mutant enzyme. This mutant protein appears to form microaggregates, which may be related to its toxicity in vulnerable neurons. Thus, the significant loss of motor neurons in transgenic mice expressing mutant SOD1 is likely to result from a toxic gain-of-function.
- SOD1 mice recapitulate many features of ALS, including axonal and mitochondrial dysfunction, progressive neuromuscular dysfunction, gliosis and motor neuron loss, an eventual death. Therefore, they have been used widely and productively to screen potential new treatments for human ALS.
Assessments
Creative Biolabs offers contract study using this transgenic mouse to test the therapeutic potential of putative new treatments for ALS. Numerous endpoints can be incorporated into a customized study design. Firstly, animals are recorded for their body weight loss, clinical observations, and survival. Moreover, grip strength, open filed test, and rotarod test can also be performed. In addition, we can conduct immunohistochemistry evaluation and MRI of the pathological alterations in response to new drugs. In brief, the assessments provided mainly include, but are not limited to:
- Body weight
- Kaplan-Meyer survival plot
- Behavioral testing (e.g., motor function, cognition, social behavior)
- Clinical observations
- Magnetic resonance imaging (MRI) for neurodegeneration in the brain and spinal cord
- Histopathological analysis
- Immunohistochemistry for motor neuron counts
The comprehensive list of rodent neurological disease models is placed below for your review:
Creative Biolabs is a premier preclinical CRO provider of in vivo pharmacology services and expert assistance in the planning, analysis, and interpretation of preclinical efficacy studies. With our years of experience and profound expertise in the field of neurological disease, we are able to deliver reliable, on-time, and quality results. Study protocols can be customized to minimize the use of resources while maximizing the information obtained.
If you are interested in our services, contact us or send us an inquiry for a formal quote. Moreover, if you are interested in other transgenic models of ALS, just contact us!
Reference
-
Wu, Shaoping, et al. "Leaky intestine and impaired microbiome in an amyotrophic lateral sclerosis mouse model." Physiological Reports 3.4 (2015): e12356. Distributed under Open Access license CC BY 4.0. The image was modified by extracting and using B part of the original image.
For Research Use Only.