Creative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsWith over a decade of experience in phage display technology, Creative Biolabs can provide a series of antibody or peptide libraries that are available for licensing or direct screening. These ready-to-use libraries are invaluable resources for isolating target-specific binders for various research, diagnostic or therapeutic applications. Highlights
Creative Biolabs has established a broad range of platforms for developing novel antibodies or equivalents. These cutting-edge technologies enable our scientists to meet your demands from different aspects and tailor the most appropriate solution that contributes to the success of your projects.
HighlightsWith deep understanding in antibody-related realms and extensive project experience, Creative Biolabs offers a variety of references to help you learn more about our capacities and achievements, including infographic, flyer, case study, peer-reviewed publications, and all kinds of knowledge that can assist your projects. You are also welcome to contact us directly for more specific solutions.
HighlightsGet a real taste of Creative Biolabs, one of the most professional custom service providers in the world. We are committed to providing highly customized comprehensive solutions with the best quality to advance your projects.
HighlightsCreative Biolabs offers customers services for protein intrinsic property modulation by computational design.
The creation of novel proteins with desired functions is a great challenge for protein engineering. We have applied computational algorithms that can search through large numbers of protein scaffolds for protein active site recapitulation, allowing rapid evaluation and testing of protein design methodologies. These methods can be directly applied to design new proteins, and provide a powerful in silico test to enhance computational protein engineering.
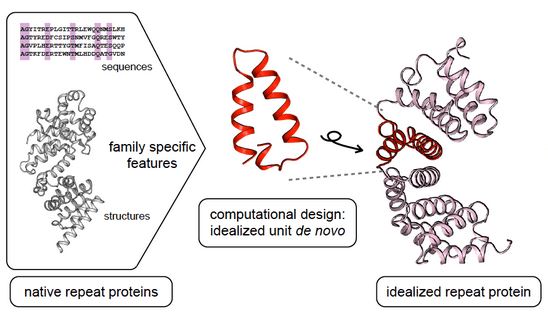
Figure 1. The general computational protein engineering approach. (Parmeggiani, F. et al., 2014)
Folding behaviors are the intrinsic properties of proteins, and there are roughly two types, as shown by protein folding experiments: the two-state kinetics and the multi-state kinetics. The determining factors for the folding type of a protein is still largely unclear, although the two types of protein folding kinetics have been known since long. A comparative study based on a collection of 43 two-state and 42 multi-state folders was performed, at different levels of proteins' intrinsic properties from the simplest sequence length to native structure topology. The protein's amino acids composition and the long-range interaction-based topological complexity were shown to determine the folding kinetics type, rather than secondary structure contents.
It is believed that all proteins are potentially allosteric, which is a consequence of re-distributions of protein conformational ensembles. The binding site shape may not show a concerted second-site change and enzyme kinetics may not reflect an allosteric transition for the supposedly non-allosteric proteins. However, a population shift can be initiated by appropriate ligands, point mutations, or external conditions.
Computationally, the 20 amino acids can be presented by a novel cylindrical representation, whereby the residue types are put in a circle and sequence positions along the z axis. In this way, the visualization of both the composition and sequence order of amino acids at is made possible at the same time. Using this model, the intrinsic properties of protein sequences are described by ten numerical descriptors and one weighted numerical descriptor. The model has been applied to conduct similarity/dissimilarity analysis of nine ND5 proteins. These numerical descriptors are proved to be more effective than several classical numerical matrices. We will work on the modulation of the intrinsic properties of your proteins with carefully chosen computational methods, and take into consideration all other factors as well.
Reference
All listed services and products are For Research Use Only. Do Not use in any diagnostic or therapeutic applications.