
Heterobifunctional degraders targeting BCR-Abl, BTK, CDK9, and DAPK1 offer a strategy to bypass kinase inhibitor resistance. Explore ligand design principles.
Introduction
Targeted protein degradation has emerged as one of the most compelling strategies in modern drug discovery, offering a fundamentally different approach to modulating disease-driving proteins. Unlike conventional small-molecule inhibitors that merely block a protein’s active site, heterobifunctional degraders redirect the ubiquitin-proteasome system to eliminate target proteins entirely. This paradigm shift from occupancy-driven to event-driven pharmacology has opened new possibilities for addressing historically challenging drug targets, particularly in oncology and neurodegeneration.
Kinase proteins represent some of the most clinically validated targets in drug development, yet resistance mutations and kinase-independent scaffolding functions frequently limit the long-term efficacy of traditional inhibitors. Heterobifunctional degraders targeting BCR-Abl, BTK, CDK9, and DAPK1 exemplify how the protein degradation approach can overcome these limitations. This article examines the scientific rationale, ligand design strategies, and practical considerations for developing kinase-targeting degraders across these four therapeutically important proteins.
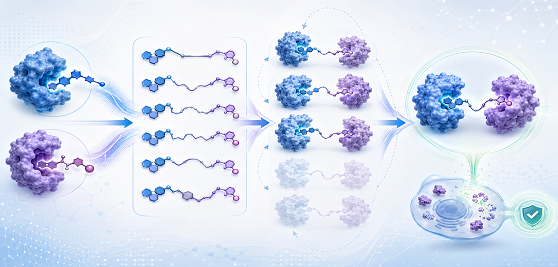
The Targeted Protein Degradation Mechanism
At the core of targeted protein degradation lies the hijacking of the endogenous ubiquiti-proteasome system, a cellular quality-control system responsible for eliminating misfolded, damaged, or surplus proteins. Heterobifunctional degraders are chimeric molecules comprising three functional elements: a target-protein-binding ligand, an E3 ubiquitin ligase-recruiting moiety, and a linker connecting the two. By simultaneously engaging both the target protein and an E3 ligase such as cereblon (CRBN) or von Hippel-Lindau (VHL), these molecules induce proximity-driven ubiquitination of the target, tagging it for proteasomal destruction.
The event-driven nature of this mechanism provides several advantages over conventional inhibition. Rather than requiring sustained target occupancy, a single degrader molecule can processively ubiquitinate multiple target copies, enabling catalytic activity at substoichiometric concentrations. Furthermore, complete protein elimination removes all functions of the target — enzymatic, scaffolding, and protein-protein interaction roles alike — a benefit that proves especially relevant for the kinase targets discussed below.
BCR-Abl-Targeting Protein Degraders
BCR-Abl, the product of a reciprocal translocation between chromosomes 9 and 22, is the hallmark oncogenic driver of chronic myeloid leukemia (CML). The development of BCR-Abl tyrosine kinase inhibitors (TKIs) has transformed CML from a fatal malignancy into a manageable chronic condition. However, only approximately one-third of patients achieve treatment-free remission, and about 10% develop primary or acquired resistance, underscoring the limitations of kinase inhibition alone.
A critical insight driving interest in BCR-Abl-targeting Protein Degraders is the recognition that BCR-Abl possesses kinase-independent scaffolding functions essential for leukemic stem cell survival. These functions — including altered DNA damage repair, aberrant cytoskeletal remodeling, and disrupted adhesion signaling — persist even under complete kinase inhibition and contribute to residual disease. By inducing complete BCR-Abl degradation, heterobifunctional degraders may simultaneously ablate both catalytic and scaffolding activities, offering a potential route toward deeper molecular remission.
Ligand design for BCR-Abl degraders leverages well-characterized TKI pharmacophores as target-binding warheads, including imatinib, dasatinib, and GZD824 derivatives. Additionally, the myristoyl-binding allosteric pocket of Abl provides an alternative binding site, enabling the development of degraders that engage BCR-Abl in its autoinhibited conformation. E3 ligase selection — whether CRBN-, VHL-, or cIAP1-based — and linker geometry optimization are critical parameters influencing degradation efficiency and selectivity.
BTK-Targeting Protein Degraders
Bruton’s tyrosine kinase (BTK) is a non-receptor tyrosine kinase of the Tec family that serves as an essential signaling node in B-cell receptor pathways. Covalent BTK inhibitors have achieved remarkable clinical success in B-cell malignancies including chronic lymphocytic leukemia and mantle cell lymphoma. Nevertheless, acquired resistance driven by BTK C481S mutations — which prevent covalent inhibitor binding — and the emergence of kinase-independent BTK scaffolding functions have prompted investigation of alternative therapeutic modalities.
BTK-targeting Protein Degraders address both resistance mechanisms by eliminating the BTK protein entirely. Studies have demonstrated that heterobifunctional degraders can achieve rapid, selective, and prolonged BTK depletion in both in vitro and in vivo models. Notably, research that characterized covalent BTK ternary complexes compatible with targeted protein degradation, providing structural insights into degrader-induced cooperativity between BTK and CRBN.
Rational ligand design for BTK degraders draws on the extensive structural biology of the BTK kinase domain, including its ATP-binding pocket, hinge region, and distal regulatory sites. The PH, SH3, SH2, and kinase domains each present opportunities for ligand engagement. Computational modeling of ternary complex formation — involving the degrader, BTK, and the chosen E3 ligase — has become an indispensable tool for optimizing linker length, attachment vectors, and degradation kinetics.
CDK9-Targeting Protein Degraders
Cyclin-dependent kinase 9 (CDK9) functions as the catalytic subunit of the positive transcription elongation factor b (P-TEFb) complex and governs the transcriptional elongation of short-lived anti-apoptotic proteins, including MCL-1 and MYC. CDK9 is overexpressed or hyperactivated across multiple malignancies — including acute myeloid leukemia, glioblastoma, breast cancer, and prostate cancer — making it an attractive therapeutic target. However, the high conservation of the ATP-binding pocket across the CDK family presents a significant challenge for developing selective ATP-competitive inhibitor.
CDK9-targeting Protein Degraders exploit differences in surface-exposed lysine residues and overall surface topology among CDK family members to achieve degradation selectivity that inhibition alone cannot provide. Even when a degrader’s target-binding ligand exhibits moderate kinase selectivity, the requirement for productive ternary complex formation with a specific E3 ligase introduces an additional selectivity filter.
Ligand design strategies for CDK9 degraders employ diverse chemical starting points. Aminopyrazole and aminothiazole scaffolds serve as core pharmacophores, while the natural product wogonin has been identified as a potent and selective CDK9 ligand suitable for degrader conjugation. Structure-based optimization of linker composition, attachment vectors, and E3 ligase choice — most commonly CRBN-based immunomodulatory drug moieties — can substantially enhance degradation potency and cellular selectivity. The ultimate goal is to achieve potent CDK9 depletion that suppresses oncogenic transcription while sparing other CDK family members essential for normal cellular function.
DAPK1-Targeting Protein Degraders
Death-associated protein kinase 1 (DAPK1) occupies a unique position among the kinases discussed here, with documented roles in both tumor suppression and neurodegeneration. DAPK1 promoter hypermethylation and consequent transcriptional silencing have been observed in nearly 30 distinct human tumor types, including lung, cervical, and gastrointestinal cancers, suggesting a tumor-suppressive function. Conversely, elevated DAPK1 expression and kinase activity in the central nervous system contribute to tau hyperphosphorylation, amyloid-beta-mediated neurotoxicity, and neuronal apoptosis — hallmarks of Alzheimer’s disease pathology.
The opposing roles of DAPK1 in cancer versus neurodegeneration demand exceptionally precise pharmacological control. DAPK1-targeting Protein Degraders offer a context-dependent strategy for eliminating DAPK1 in neurological disease while potentially preserving its expression in tumor contexts where its pro-apoptotic functions are desirable.
Ligand design for DAPK1 degraders benefits from the protein’s multi-domain architecture, which includes kinase, calmodulin-binding, ankyrin repeat, and death domains. Computer-aided drug design approaches — including fragment-based ligand discovery, molecular docking, and binding free-energy calculations — facilitate the identification of ligands that engage the ATP-binding pocket, allosteric sites, or substrate-binding interfaces. Both ATP-competitive and non-ATP-competitive ligands have been characterized, including the high-affinity inhibitors TC-DAPK 6 and HS-38. Peptide ligands derived from the DAPK1 substrate p21, engineered antibody-based binders identified through surface plasmon resonance and nuclear magnetic resonance screening, and natural product scaffolds modified via medicinal chemistry each provide viable starting points for degrader development.
Key Considerations in Degrader Ligand Design
Designing effective heterobifunctional degraders requires balancing multiple interdependent parameters. The target-binding ligand must exhibit sufficient affinity and selectivity — though interestingly, degrader selectivity can exceed that of the ligand alone, as productive ternary complex formation imposes additional structural constraints. Linker composition, length, and rigidity profoundly influence degradation efficiency by modulating target-ligase proximity, ternary complex stability, and the spatial orientation of surface lysines available for ubiquitination.
E3 ligase selection represents another critical decision point. CRBN-recruiting immunomodulatory drug moieties offer favorable drug-like properties and oral bioavailability, while VHL-based recruiters provide a well-characterized structural framework for rational design. The choice between these and other E3 ligases — including cIAP1 and MDM2 — depends on target-specific factors such as lysine distribution, tissue expression patterns of the ligase, and the desired degradation profile.
Additional practical considerations include the cell permeability and metabolic stability of the degrader molecule. Heterobifunctional degraders often exceed the molecular weight of conventional small-molecule drugs, placing them in the beyond-Rule-of-5 chemical space. Consequently, early assessment of solubility, permeability, and metabolic stability can help prioritize degrader candidates with favorable developability profiles. For research teams navigating these complexities, partnering with an experienced preclinical contract research organization (CRO) can streamline degrader discovery workflows and provide access to specialized ligand design, linker chemistry, and degradation assay capabilities.
Integrated Protein Degrader Development Support
Translating a protein degradation hypothesis into validated degrader candidates requires coordinated expertise spanning medicinal chemistry, structural biology, biochemical assay development, and cellular pharmacology. Creative Biolabs offers comprehensive protein degrader ligand design services that support research teams at every stage of the discovery process, from target feasibility assessment through lead optimization.
The ligand design platform accommodates diverse chemical starting points, including small-molecule inhibitors, peptide ligands, antibody-derived binders, and natural product scaffolds. Each project benefits from a tailored experimental design that aligns with the specific biochemical characteristics of the target protein and the research objectives of the sponsoring team. Computer-aided drug design workflows — encompassing molecular docking, binding free-energy prediction, and ternary complex modeling — guide rational optimization of ligand affinity, selectivity, and degrader-compatible exit vectors.
For research programs requiring integrated support, Creative Biolabs provides end-to-end services including ligand synthesis, linker chemistry, E3 ligase recruiter conjugation, biochemical binding assays, cellular target engagement studies, and degradation kinetic analysis. Flexible engagement models ensure alignment with the scope and stage of each project, whether the goal is early-stage proof-of-concept or advancement toward lead nomination.
Conclusion
Heterobifunctional degraders targeting BCR-Abl, BTK, CDK9, and DAPK1 illustrate the transformative potential of targeted protein degradation across oncology and neurodegeneration. By eliminating proteins rather than merely inhibiting them, these molecules address resistance mechanisms and scaffolding functions that limit conventional kinase inhibitors. Successful degrader development hinges on thoughtful ligand design — encompassing target-binding warhead selection, linker optimization, and E3 ligase pairing — supported by rigorous biochemical and cellular characterization.
For research teams pursuing protein degrader programs, access to specialized chemistry and pharmacology capabilities can accelerate progress from concept to candidate. Contact the scientific team at Creative Biolabs to discuss tailored degrader design strategies for your kinase target of interest.
FAQ
What is targeted protein degradation?
Targeted protein degradation is a therapeutic strategy that uses heterobifunctional molecules to redirect the ubiquitin-proteasome system toward disease-relevant proteins. These chimeric molecules simultaneously bind a target protein and an E3 ubiquitin ligase, bringing them into proximity such that the target becomes ubiquitinated and subsequently degraded by the proteasome. Unlike conventional inhibitors that block a single functional site, degradation eliminates the entire protein and all its associated activities.
Why are kinase proteins attractive targets for heterobifunctional degraders?
Kinases are among the most extensively validated drug targets, but resistance mutations and kinase-independent scaffolding functions frequently limit the long-term effectiveness of ATP-competitive inhibitors. Heterobifunctional degraders can overcome both challenges: by eliminating the entire protein, they ablate scaffolding functions that persist under kinase inhibition, and they remain effective against many resistance mutations that disrupt inhibitor binding without impairing degrader-induced ubiquitination.
How is degrader selectivity achieved for kinases with highly conserved ATP-binding pockets?
Degrader selectivity arises from two sources. First, even a moderately selective target-binding ligand can impart selectivity. Second, productive degradation requires formation of a stable ternary complex involving the degrader, the target, and the E3 ligase — and the protein-protein interface formed in this complex introduces an additional selectivity filter. Differences in surface lysine distribution, protein surface topology, and ternary complex geometry among related kinases can yield degradation selectivity exceeding the binding selectivity of the ligand per se.
What are the main challenges in designing heterobifunctional degraders?
Key challenges include optimizing the linker — its length, composition, and attachment points — to achieve efficient ternary complex formation without destabilizing protein-protein interactions; selecting an appropriate E3 ligase based on target lysine distribution and tissue expression; achieving favorable drug-like properties given that degraders often fall in beyond-Rule-of-5 chemical space; and avoiding off-target degradation of proteins structurally related to the intended target.
How can partnering with a preclinical CRO support protein degrader discovery programs?
An experienced preclinical CRO can provide integrated support spanning ligand design, medicinal chemistry, linker optimization, E3 ligase recruiter conjugation, biochemical binding assays, cellular target engagement studies, and degradation kinetic analysis. This enables research teams to access specialized capabilities without building internal infrastructure, accelerating the transition from target validation to lead degrader nomination.