The complement system has long been regarded as a fundamental component of innate immunity, with its three well-established activation routes—classical, lectin, and alternative pathways—serving as canonical pillars in immunology textbooks. However, a groundbreaking study by Dr. Michael B. Brenner’s team at Harvard Medical School, published in Nature (2025), introduces a paradigm-shifting concept: Granzyme K (GZMK), a serine protease secreted by CD8+ T cells, can independently initiate a full complement cascade. This fourth, previously unrecognized pathway opens new horizons in the understanding and therapeutic targeting of immune-mediated diseases.
Complement and Cancer
Traditionally, Granzyme family members such as GZMA and GZMB were known for inducing apoptosis via perforin-mediated entry into target cells. However, recent studies have indicated that GZMK can be secreted extracellularly, particularly in inflamed tissues, suggesting a non-canonical, perforin-independent role in immune modulation.
The research team observed persistent extracellular GZMK secretion by both resting and activated CD8+ T cells, with significantly elevated levels in the synovial tissue of rheumatoid arthritis (RA) patients. This led to a pivotal question: What function does extracellular GZMK serve in the disease microenvironment?
Structural Clues: GZMK Resembling Complement Serine Proteases
To explore GZMK’s potential targets, the team conducted a structural comparison between GZMK and known complement proteases. Surprisingly, the catalytic domain of GZMK closely resembled that of complement factor D (CFD), though GZMK failed to cleave CFD’s substrate. Shifting focus, they uncovered structural similarities with C1s and MASP1, key initiators of the classical and lectin pathways.
This similarity prompted a hypothesis: Could GZMK functionally substitute for C1s or MASP1 in initiating complement activation?
Dissecting the Complement Cascade Initiated by GZMK
The team evaluated GZMK’s capacity to recapitulate the three core phases of complement activation—recognition, initiation, and execution—through rigorous in vitro assays and in vivo models.
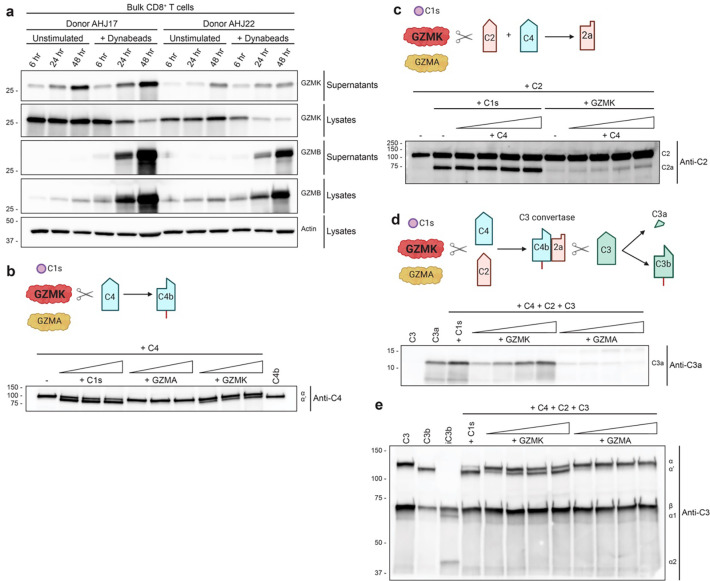
Fig. 1 GZMK cleaves the complement components C4 and C2 to generate a C3 convertase that cleaves C3 into C3a and C3b.1,2
- Recognition: Heparan Sulfate Receptor-Dependent Membrane Anchoring
Unlike classical activation, which is antibody-dependent, GZMK was shown to bind the cell surface via heparan sulfate receptors. Blocking these receptors abrogated GZMK localization and subsequent complement activation, revealing a unique Recognition mechanism independent of immune complexes.
This suggests therapeutic potential in blocking GZMK-heparan interactions to modulate complement-driven inflammation.
- Initiation: Formation of C3 Convertase (C4b2b)
The Initiation phase was experimentally dissected:
- C4 cleavage: GZMK, unlike GZMA, could cleave C4 into C4a and C4b.
- C2 cleavage: GZMK facilitated the formation of C2b.
- C3 activation: When incubated with C4 and C2, GZMK enabled the formation of C4b2b, which subsequently cleaved C3 into C3a and C3b.
GZMK does not cleave C3 directly. C3 activation is contingent on the prior formation of a C3 convertase.
- Execution: Terminal Effector Functions via MAC Formation
In the Execution phase:
- C3b aided in assembling the C5 convertase, initiating C5 cleavage.
- C5a, a potent anaphylatoxin, and C5b, a precursor for membrane attack complex (MAC), were generated.
- GZMK activation-induced mast cell degranulation and complement-mediated cell lysis, confirming full cascade execution.
GZMK initiates and drives a complete complement response independently of C1q, antibodies, or mannose-binding lectins.
A Fourth Pathway of Complement Activation
By fulfilling all functional checkpoints—Recognition (via heparan binding), Initiation (via C4/C2 cleavage), and Execution (via C3/C5 processing)—GZMK meets the criteria of a bona fide complement activation pathway.
This discovery proposes a fourth complement activation pathway, distinct from classical, lectin, and alternative pathways, with CD8+ T cells and GZMK as central initiators.
To evaluate the clinical relevance of this pathway, researchers examined RA synovial tissue and conducted murine disease models:
- Fibroblast response: Inflammatory cytokines (e.g., IFN-γ, TNF-α) upregulated C3/C4 expression in synovial fibroblasts, increasing substrate availability for GZMK.
- Feedback loop: A CD8+ T cell-fibroblast axis amplifies local complement activation.
- GZMK knockout models: Mice lacking GZMK showed reduced severity of arthritis and psoriatic inflammation, with lower C3a and C5a levels in lesions.
- Human data: Serum GZMK levels in RA patients positively correlated with C3a/C5a concentrations.
Therapeutic Outlook and Future Directions
The identification of GZMK as a novel complement activator unveils several therapeutic avenues:
- GZMK secretion: Inhibitors of T cell-derived GZMK
- Heparan binding: Receptor antagonists to block membrane anchoring
- Proteolytic activity: Small-molecule GZMK inhibitors
- Substrate limitation: C4/C2 expression regulation
At Creative Biolabs, we offer comprehensive complement profiling services, including:
- Quantification of C3a, C5a, and MAC formation
- Complement activation pathway mapping
Learn more about our complement activation assay platforms.
The GZMK-mediated complement activation route represents a paradigm shift in immunology. It not only enriches our understanding of T cell effector mechanisms but also offers novel insights into the etiology of autoimmune diseases. Future research exploring GZMK inhibitors, heparan-blocking agents, and substrate modulation may pave the way for targeted therapeutics in RA, psoriasis, and beyond.
As complement biology enters a new era, Creative Biolabs stands ready to support your innovation with tailored assays and discovery solutions.
References
- Donado, Carlos A., et al. “Granzyme K drives a newly-intentified pathway of complement activation.” bioRxiv (2024). https://doi.org/10.1101/2024.05.22.595315
- Distributed under Open Access license CC BY 4.0, without modification.