Cancer is a complex, multifactorial disease that evolves through a series of functional capabilities, widely known as the hallmarks of cancer. Among the numerous biological systems influencing these hallmarks, the innate immune complement system has emerged as a critical and surprisingly multifaceted player. Traditionally recognized for its role in immune defense against pathogens, complement proteins now show tumor-promoting and tumor-suppressing activities, operating not only through canonical pathways but also via non-canonical, intracellular mechanisms.
At Creative Biolabs, we are at the forefront of complement system research, providing comprehensive solutions for complement analysis and complement-targeted therapeutic development. This blog explores how complement proteins interact with the hallmarks of cancer, offering novel therapeutic opportunities.
Complement and Cancer
The complement system comprises over 30 plasma and membrane-bound proteins that function as part of the body’s first line of defense. It recognizes pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), orchestrating immune responses through:
- Opsonization (coating of pathogens for phagocytosis)
- Inflammation (via anaphylatoxins C3a and C5a)
- Complement-dependent cytotoxicity (CDC) (via membrane attack complex, MAC)
However, tumors often exploit complement regulatory mechanisms to evade destruction. By overexpressing complement inhibitors like CD46, CD55, and CD59, cancer cells escape complement-mediated lysis while sustaining an inflammatory tumor microenvironment (TME) conducive to tumor progression.
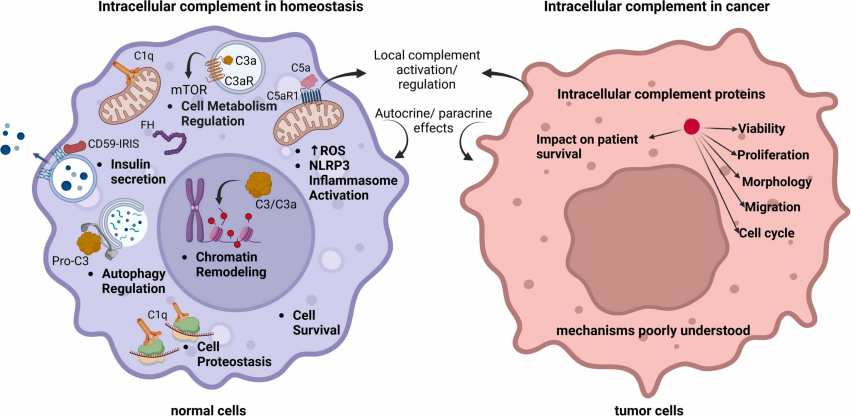
Fig. 1 Intracellular complement in homeostasis and cancer.1,2
Recent research reveals that complement proteins:
- Act intracellularly (forming the “complosome”) to regulate metabolism, survival, and cell cycle.
- Function extracellularly outside of cascade-dependent activation.
- Interact with non-complement molecules, influencing transcription, autophagy, and immune modulation.
This expanded view transforms the complement from a simple cascade into a dynamic system impacting nearly every hallmark of cancer.
How Complement Contributes to Cancer Hallmarks
- Tumor-Promoting Inflammation
Complement activation within tumors releases C3a and C5a, recruiting myeloid-derived suppressor cells (MDSCs) and promoting chronic inflammation. Rather than eliminating cancer, this environment favors immune suppression, angiogenesis, and genetic instability. Inhibiting C5aR1, particularly in combination with checkpoint blockade, has shown synergistic anti-tumor effects.
Creative Biolabs Solutions: We provide C3a/C5a quantification assays and complement inhibition screening to support anti-inflammatory therapeutic strategies.
- Avoiding Immune Destruction
While MAC formation can kill tumor cells, most malignancies evade this via overexpressing regulators (e.g., CD55, CD59, Factor H). Furthermore:
- iC3b on tumor surfaces engages CR3 on macrophages, inducing tolerogenic responses.
- C3d, in contrast, may act as an immunogenic adjuvant—enhancing antigen presentation and T-cell activation.
This duality offers novel targets for immunotherapy, including engineered anti-C3d antibodies or FH inhibitors.
Creative Biolabs Solutions: We offer complement regulator profiling services and anti-tumor MAC formation assays to accelerate immunotherapy development.
- Resisting Cell Death
Sublytic MAC deposition can paradoxically activate survival pathways (e.g., via ERK1/2 and AKT), helping tumor cells resist apoptosis and necrosis. Additionally, intracellular complement proteins such as C1q, C1s, and Factor H modulate anti-apoptotic signaling independently of cascade activation.
- Sustaining Proliferative Signaling
Complement anaphylatoxins stimulate proliferation by activating pathways like:
- PI3K/Akt (e.g., C5aR1 signaling)
- β-catenin (e.g., C3aR1 activation)
Intracellular C3aR1 and C5aR1 interactions stabilize transcription factors and promote oncogenic gene expression. Silencing complement components like C1s or Factor H significantly impair cell growth in RCC and SCC models.
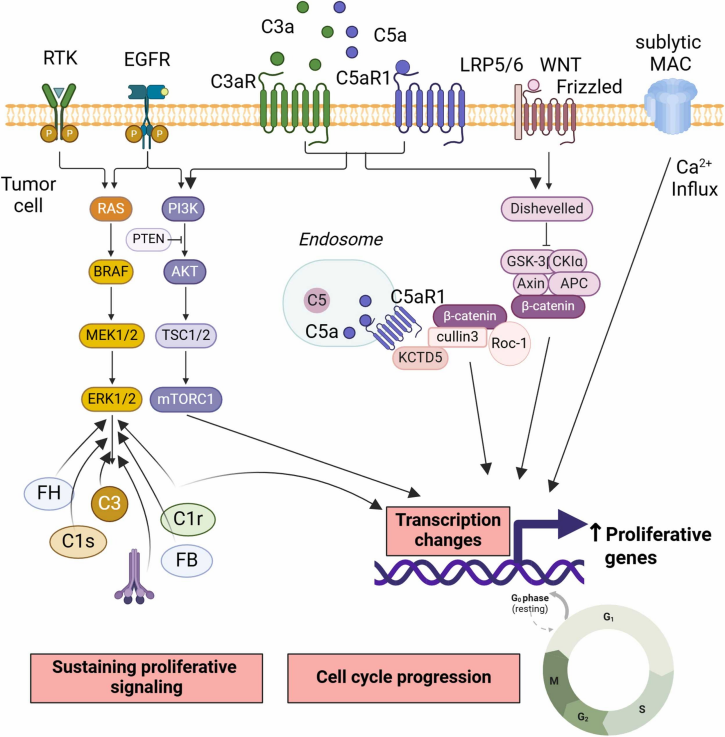
Fig. 2 Complement sustains proliferative signaling.1,2
- Inducing Invasion and Metastasis
Complement molecules drive tumor invasion and metastasis by:
- Promoting epithelial-mesenchymal transition (EMT).
- Enhancing matrix metalloproteinases (MMPs) expression.
- Disrupting vascular barriers via C3a/C5a-mediated neutrophil extracellular trap (NET) formation.
EVs enriched with Factor H or C3 also contribute to pre-metastatic niche formation—targets now being explored via anti-FH therapeutic antibodies.
- Angiogenesis Promotion
Complement components, especially C5a and C1q, promote tumor angiogenesis:
- C3a and C5a directly activate endothelial cells and indirectly promote VEGF secretion by tumor-associated macrophages.
- C1q also drives vessel formation independently of cascade activation, reinforcing its context-dependent angiogenic role.
- Shaping Cellular Metabolism
Acidification of the TME enhances complement activation, potentially sustaining the Warburg effect in cancer cells. Acidic pH from glycolytic metabolism also enhances complement activation, shaping immune and metabolic dynamics in the TME. Intracellular complement proteins also regulate key metabolic pathways critical for tumor survival and progression.
- Facilitating Immune Evasion and Epigenetic Reprogramming
Intracellular complement influences epigenetics through:
- METTL3-mediated m6A modifications (via C5aR1)
- Nuclear C3b complexing with SIN3A/HDACs to repress tumor suppressor genes (e.g., GADD45A)
- Non-coding RNAs like circ-CFH modulating miRNA availability to upregulate oncogenes
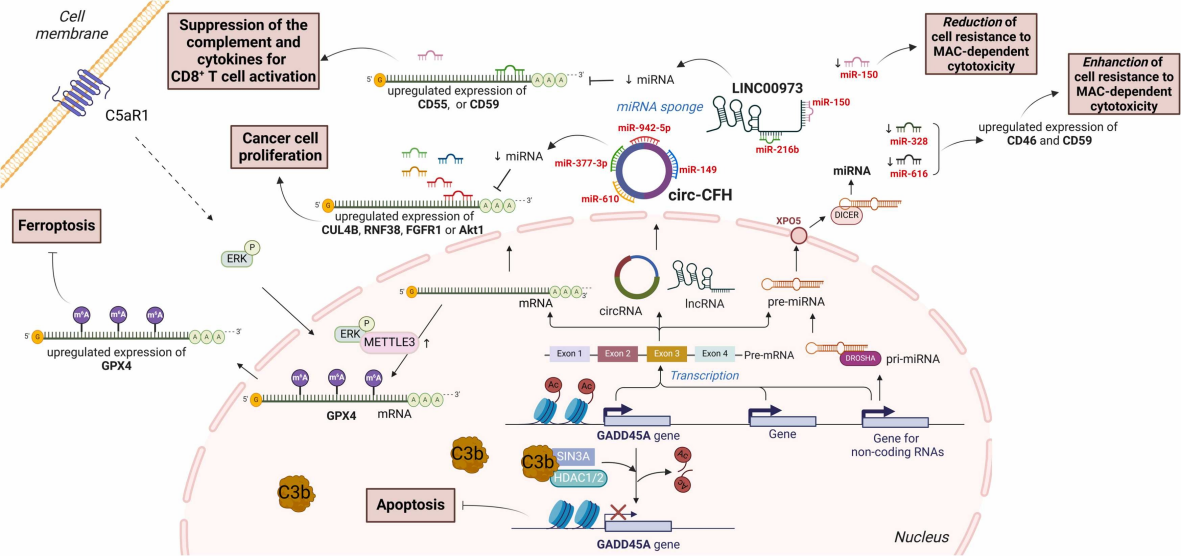
Fig. 3 The role of complement in epigenetic reprogramming in tumors.1,2
- Impacting Genome Stability and Tumor Microbiome
Complement-driven chronic inflammation leads to DNA damage and mutation accumulation. Additionally, microbiota-driven complement activation can promote or inhibit cancer progression, depending on the microbial composition and context. Complement crosstalks with the tumor microbiota, as shown by MBL-mediated activation in response to fungi like Malassezia.
Complement activation may initially support immune-mediated tumor elimination. However, as tumors evolve, complement can become an accomplice, driving inflammation, immune evasion, and metastasis. Given the context-dependent roles of complement, targeted complement therapies must be carefully designed, and ideally personalized based on tumor-specific complement profiles.
Translating Discovery into Therapy
Given its duality, targeting the complement system in cancer is nuanced. Inhibiting specific components (e.g., C5aR1, FH) may benefit some tumor types, while others may require enhancing complement-mediated cytotoxicity.
At Creative Biolabs, we provide cutting-edge solutions to dissect and modulate the complement system:
- Complement Pathway Profiling—Comprehensive analysis of classical, lectin, and alternative pathways
- Complement Profiling Panels—Quantify C3a, C5a, MAC, and regulators in tumor samples
- FH/C1q Functional Assays—Evaluate complement evasion mechanisms and immune suppression
- Cellular Localization of Complosome—Visualize intracellular complement components in cancer cell models
- Complement-Driven Therapy Screening—Test synergy with checkpoint inhibitors, radiotherapy, or targeted antibodies
Toward Complement-Based Cancer Immunotherapies
Complement is not just an ancient defense cascade—it’s a multifaceted modulator of tumor biology. Whether as a driver of immune evasion or a trigger of immunogenic death, its roles are shaped by tumor type, cellular context, and microenvironmental cues. Understanding its dualistic functions—both extracellular and intracellular—is essential for designing next-generation complement-targeted therapies.
By integrating cutting-edge research with advanced analytical platforms, Creative Biolabs stands ready to help you uncover the full therapeutic potential of complement modulation in cancer treatment.
References
- Artero, Mikel Rezola, et al. “Complement and the hallmarks of cancer.” Seminars in Immunology. 78. Academic Press, 2025. https://doi.org/10.1016/j.smim.2025.101950
- Distributed under Open Access license CC BY 4.0, without modification.