The complement system is recognized as one of the mechanisms by which antibody drugs function. However, early antibody drug design efforts rarely focused on enhancing complement activation. As research has progressed, strategies to boost complement activation have become a key part of antibody drug design.
Based on the theory of complement activation, strategies to enhance the complement activation ability of antibodies include increasing antibody oligomerization, enhancing C1q recruitment, antibody structural hybridization, designing antibody and bystander combinations, or overcoming the inhibitory effects of complement regulatory proteins.
Enhance Cross-linking of Fc Structures between Antibodies
Antibody cross-linking is crucial for the activation of complement system. IgG or IgM antibodies activate the classical complement pathway by binding C1q via their Fc regions. C1q consists of six collagen-like arms, each containing an N-terminal triple helix and a C-terminal immunoglobulin-binding globular head.
The affinity of the individual C1q globular heads for immunoglobulin Fc is very low, so physiological binding and activation require multivalent, high-affinity interactions.
Certain mutations in the Fc region, such as E345R or the triple mutant RGY (E345R, E430G, and S440Y), promote IgG1 oligomerization, significantly enhancing complement recruitment and CDC.
E345 and E430 are hotspots controlling IgG hexamerization. Selecting the E430G and E345K mutations allows antibodies to induce IgG1 aggregation exclusively upon cell surface antigen binding while retaining the pharmacokinetic and developability characteristics of conventional antibodies.
In addition to E430 and E345 being the most common mutation hotspots, studies have also reported that the H429F mutation can enhance targeted CDC, comparable to the adjacent E430G substitution.
Benefits of IgG1-FcRn Mutations
The site of the Fc+Fc cross-linking interface mentioned above is actually located within the FcRn-antibody binding site. Therefore, mutations implemented to modulate antibody half-life may inadvertently affect IgG1 oligomerization and complement binding.
Of two validated half-life-extending mutations—YTE (M252Y/S254T/T256E) and LS (M428L/N434S)—YTE reduces C1q binding and CDC activity, while LS maintains wild-type-like behavior.
Thus, by exploiting the structural overlap between the Fc-FcRn and Fc-Fc interfaces, it is possible to specifically combine half-life extension with complement-enhancing properties. An example of this is the reported REW (Q311R/M428E/N434W) mutation in the Fc structure.
Based on the Natural Properties of IgM
IgM’s inherent multimeric properties offer another potential source of inspiration for engineered antibodies that enhance complement activation.
Using IgM alone as an antibody drug would pose significant challenges to drugability due to its relatively short half-life and poor developability. However, as an alternative, incorporating a structure that mimics IgM’s multimeric properties into the IgG1 structure (by fusion of the IgM tail to the C-terminus) could circumvent the druggability limitations of native IgM. However, while C-terminal fusion of the IgM tail results in spontaneous covalent multimerization of IgG1 (driven by the penultimate cysteine residue in the tail), it can also lead to off-target complement activation and reduced in vivo activity.
To address this issue, the second cysteine residue in the tail can be mutated (C575S). This structure significantly enhances C1q recruitment and CDC compared to conventional IgG1.
Enhance C1q binding
One approach to improving complement activation is to introduce mutations into the Fc-C1q binding sites. These sites are within or near the FG, BC, and DE loops of the CH2 structure, which are involved in the Fc-C1q interaction.
However, an interesting point is that there is overlap between the C1q and FcγR binding sites on IgG1, which may impair FcγR-mediated ADCC. For example, while the K326W mutation enhances C1q binding, it significantly impairs ADCC. Therefore, further exploration is needed in this area, such as simultaneously introducing FcγR affinity-enhancing mutations to restore or even improve ADCC.
Bispecific C1q Adapter
Bispecific C1q engagers utilize a TCE-like structure, which links C1q to cell surface antigens via a single antibody, thereby activating complement and effectively lysing target cells. This bispecific antibody offers an alternative complement recruitment method that does not require antibody clustering and Fc-C1q interactions.
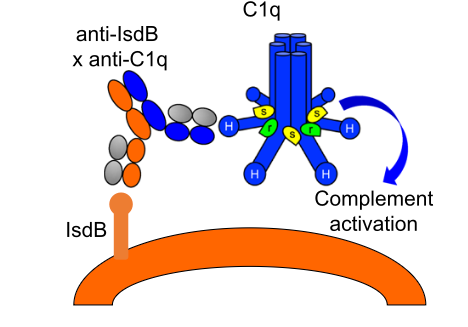
Fig. 1 Proposed mechanism of action of the anti-IsdB x anti-C1q antibody.1,2
IgG Subtype-based Manipulation
There are four IgG subtypes, of which IgG1 and IgG3 are considered the most efficient recruiters of the classical pathway. Although IgG3 has a stronger affinity for C1q and recruits C1q more efficiently, there are conflicting reports regarding the effectiveness of IgG1 and IgG3 in inducing CDC.
Generally speaking, the difference may depend on antigen density, with IgG3 and IgG1 exhibiting superior complement activation at low and high antigen densities, respectively.
The higher activity of IgG3 at low antigen levels is attributed to its long, flexible hinge, which allows it to access sparsely distributed antigens with less spatial restriction. Nevertheless, incorporating chimeric structures of IgG3 and IgG1 into antibody design can be useful for activating CDC.
In practice, however, despite the potential of IgG3 and its chimeric forms, this subclass is largely absent from clinical development. This may be due to several factors, including the lack of Protein A binding, the risk of immunogenicity due to inter-individual allotypic variation, and allotype-specific concerns regarding half-life and in vivo stability.
Other Technologies
Monoclonal antibodies have limited ability to induce CDC because monoclonal IgG antibodies bind to a single epitope on an antigen. However, studies have shown that pairs of monoclonal antibodies that do not induce CDC individually acquire this ability when used together, as they target different epitopes on the same antigen. This suggests that clonal cocktails may be an attractive strategy for enhancing therapeutic efficacy and restoring complement-dependent effector functions.
Furthermore, given that the complement system is tightly regulated by both membrane-bound and soluble factors, designing strategies to overcome complement inhibition is also a viable approach to enhancing complement recruitment.
Evidence suggests that inhibition of mCRP (CD46, CD55, and CD59) using blocking antibodies improves rituximab-induced CDC. Among mCRP, CD55 is a particularly attractive therapeutic target because it inhibits complement activation at the level of the C3 and C5 convertases. In contrast, CD46 depletion results in less potent enhancement of CDC than CD55 and CD59. In addition to mCRP blockade, regulation mediated by factor H, a soluble inhibitor that binds to anionic structures on the cell surface and inactivates cleaved C3, could also be targeted.
The structure of factor H consists of 20 homologous short consensus repeats (SCRs) arranged in a “beads-on-a-string” pattern. SCRs 19-20 play a dual role in binding to the cell surface and inactivating C3b. Recombinant SCR19-20 domains have been shown to reduce factor H binding to cells, thereby abolishing its inhibitory function and sensitizing cells to rituximab and ofatumumab.
References
- Cruz, Jonathan W., et al. “A novel bispecific antibody platform to direct complement activity for efficient lysis of target cells.” Scientific Reports1 (2019): 12031.https://doi.org/10.1038/s41598-019-48461-1
- Distributed under Open Access license CC BY 4.0, without modification.