
Conditionally activated antibody prodrug conjugate (PDC)
Traditional ADCs not only target receptors expressed on tumor cells but also those on certain healthy tissues, leading to inevitable off-tumor toxicity. Probody Drug Conjugates (PDCs) can address this issue (Figure 3), inspired by the concept of prodrugs in small molecule therapeutics. The precursor antibody molecules in PDCs are IgGs that are either fused at their N-terminus with a cleavable linker to a self-masking group or designed with antigen-binding sites that undergo pH-dependent conformational changes, thereby reducing the IgG’s affinity for its target. Upon reaching the tumor microenvironment, these antibodies either shed their self-masking groups or undergo a change in the conformation of their antigen-binding sites in response to specific tumor-associated factors (such as protease abundance and acidic conditions), thereby locally restoring their original target-binding affinity and payload release. This innovative approach holds the promise of enhancing the therapeutic index of ADCs.
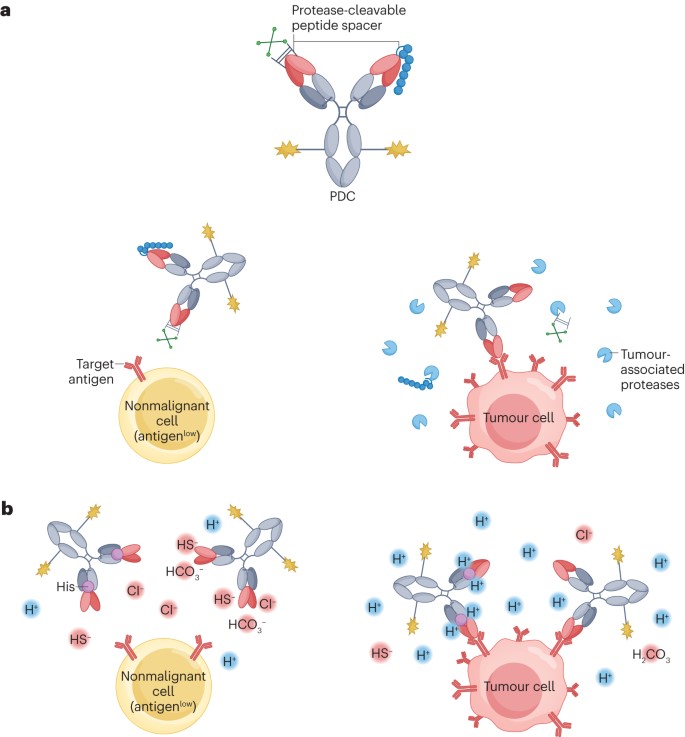
Figure 3. Conditionally activated antibody prodrug conjugates
- PDC with a protease sensitive self-masking group
A study showed that the anti-CD71 PDC CX-2029 and its ADC equivalent (both containing the MMAE payload, DAR~2) demonstrated comparable levels of antitumor activity in various solid tumor mouse xenograft models. The preclinical maximum tolerated doses (MTDs) for the PDC and its equivalent ADC in non-human primates were 6 and 0.6 mg/kg, respectively, suggesting that the introduction of the masking fragment increased the therapeutic index by approximately tenfold. Building on this concept, researchers have developed several first-in-class PDCs, including CX-2051 (Phase I clinical trials), praluzatamab ravtansine (Phase II clinical trials), and CX-2029 (Phase I/II clinical trials). Praluzatamab ravtansine contains the microtubule inhibitor DM4 and an anti-CD166 antibody. CD166 is broadly expressed in many non-malignant tissues, making it an ideal target for a PDC. In a Phase I clinical trial, 99 patients with metastatic solid tumors, comprising breast cancer (46%), epithelial ovarian cancer (22%), and non-small cell lung cancer (13%), were treated with praluzatamab ravtansine, with tumor regression observed at doses ≥4mg/kg. CX-2029, an anti-CD71 PDC with an MMAE payload, is currently undergoing Phase I clinical trials involving patients with various advanced solid tumors, such as NSCLC (20%), HNSCC (18%), and colorectal cancer (16%).
- PDC with a pH reactive antigen binding site
Tumor tissues (pH = 6.0–6.8) are typically slightly more acidic than most healthy tissues (pH = 7.3–7.4). The difference in pH values can be utilized for conditional activation of ADCs due to reversible conformational changes in the antigen-binding sites (Figure 3b). Incorporating weakly basic histidine residues into the antibody’s binding region is a common method to impart this pH-dependent activation. A prime example is the MMAE-based EGFR-targeting PDC, HTI-1511. The parent antibody of HTI-1511 exhibits more than tenfold higher binding affinity to EGFR at pH = 6.0–6.5 compared to pH = 7.4. HTI-1511 demonstrated good tolerability up to a dose of 8 mg/kg in cynomolgus monkeys, indicating potential clinical safety. In conclusion, the PDC platform holds significant promise for targeting antigens, yet the types of specific cancer subtypes effectively targeted by PDCs still need to be determined.
Immunostimulatory ADC (ISAC)
ISACs enhance the immune response against tumors by activating the immune system. Compared to traditional ADCs that carry cytotoxic payloads, ISACs offer the following potential advantages:
- ISAC-mediated antitumor responses can target a variety of tumor-associated damage-associated molecular patterns (DAMPs).
- ISAC-mediated immune stimulation can activate not only antigen-presenting cells but also other tumor-infiltrating immune cells, such as T cells.
- ISACs can induce immune memory effects that span the entire cellular immune response, thereby providing lasting antitumor effects and reducing the risk of recurrence.
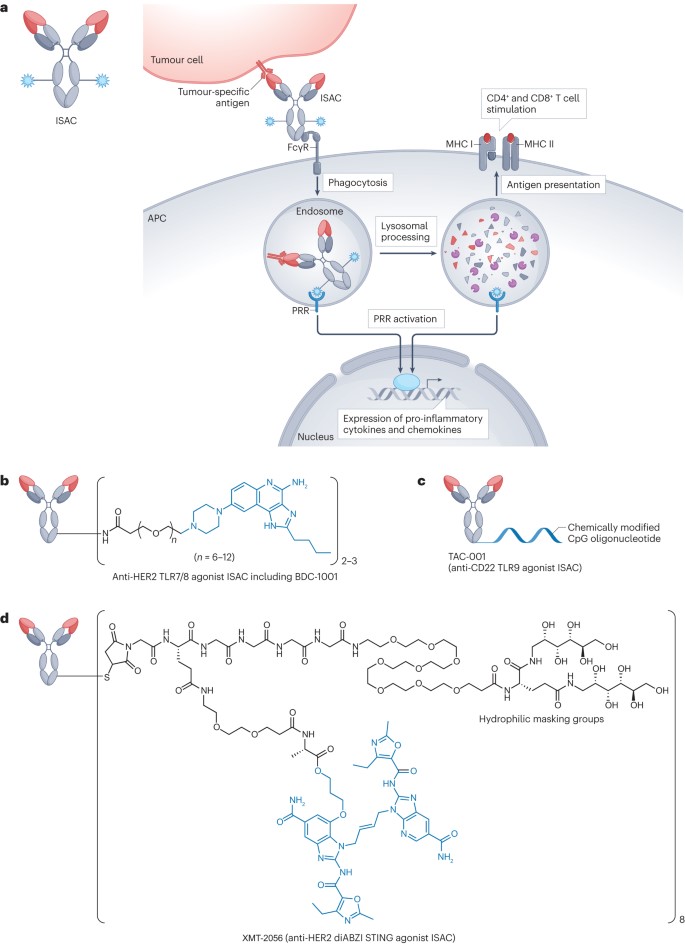
Figure 4. Immunostimulatory ADCs
- ISAC with TLR7/TLR8/TLR9 agonist payload
Among all TLRs identified so far, TLR7, TLR8, and TLR9 are the primary targets for most ISACs, with the activation of these endosomal TLRs promoting the presentation of tumor-associated DAMPs by APCs, generating robust antitumor effects through the activation of both innate and adaptive immune responses. In 2015, researchers announced the activity of an anti-CD20 ISAC developed by combining a TLR7 agonist with rituximab. In vitro tests showed that the combination did not impair the antigen-binding ability or specificity of rituximab, nor did it affect the TLR-stimulating activity of the agonist. This early study paved the way for the development of various ISACs activating TLR7/8. However, in the Phase I study (NCT04460456) and Phase I/II study (NCT05091528) of the HER2 TLR8 ISAC SBT6050, cytokine-related adverse events ultimately led to the termination of the study. Another ISAC targeting HER2, NJH395, containing a TLR7 agonist linked to an anti-HER2 antibody via a non-cleavable linker, was evaluated in a Phase I trial (NCT03696771). However, the study was also stopped after completing the single ascending dose part due to insufficient efficacy, among other reasons. Another HER2-targeting ISAC, BDC-1001, is a rituximab-based ISAC equipped with a TLR7/8 agonist via a non-cleavable linker (Figure 4b). BDC-1001 is currently being tested in a Phase I/II trial. Unlike SBT6050 and NJH395, BDC-1001 has shown promising preliminary results.
- ISAC with STING agonist payload
When exposed to exogenous DNA from microbial pathogens and/or dying tumor cells, the cGAS-cGAMP-STING pathway is activated, leading to the production of type I interferons and the activation of innate immunity. Research has shown that STING signaling is critical for inducing T-cell-mediated antitumor immune responses and the infiltration of T cells into the tumor microenvironment (TME). An EGFR-targeting ISAC conjugating the cGAMP analog IMSA172 to an anti-EGFR antibody demonstrated good tolerability in EGFR-expressing mouse xenograft models, with dosing every three days for three doses at 200μg (approximately 8-10mg/kg), showing promising antitumor activity. The antitumor activity of these ISACs was further enhanced when combined with anti-PD-L1 antibodies. In summary, the multimodal antitumor immune actions provided by ISACs show potential for reducing the risk of acquired resistance and/or delaying its onset. With their unique mechanism of action, ISACs hold promise as a powerful tool for addressing the complex immune landscape in cancer treatment.
Protein degradation ADC (DAC)
DACs leverage the cell’s protein degradation mechanisms to target specific proteins, offering an innovative approach to cancer treatment. This design concept is derived from PROTAC technology. PROTACs are heterobifunctional molecules connected by a linker, consisting of two ligands where one ligand targets a specific protein of interest and the other ligand binds to an E3 ubiquitin ligase. Unlike traditional inhibitors, the binding of these ligands to the target protein does not require antagonistic activity. Such a design enables the targeting of proteins previously considered “undruggable” as potential therapeutic targets.
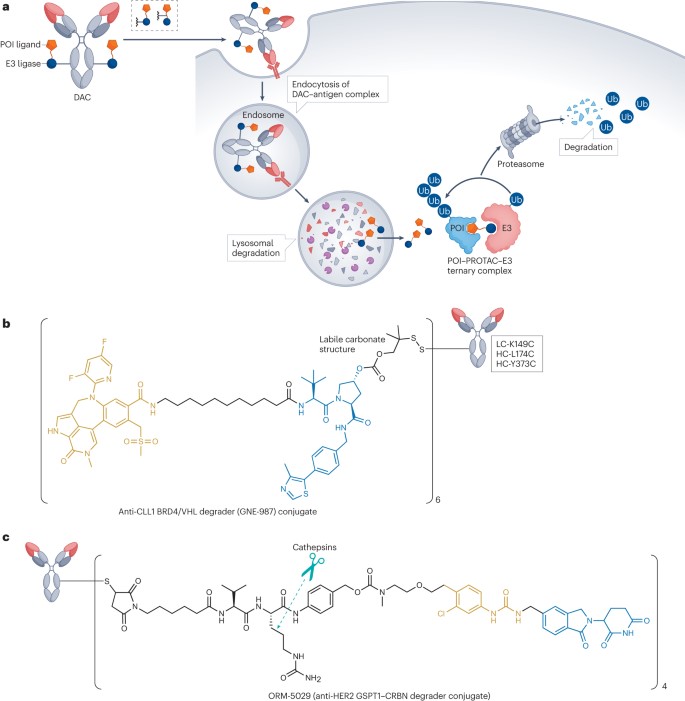
Figure 5. Protein degradation of ADCs
GNE-987 is a BET-PROTAC comprised of a BRD4 ligand and a VHL ligand, demonstrating effective degradation of BRD4 (DC50 = 0.03 nM) in EOL-1 cells and showing exceptional in vitro potency in two cell lines (IC50 = 0.02 nM in EOL-1 and IC50 = 0.03 nM in HL-60). However, drug metabolism and pharmacokinetics rendered it ineffective in in vivo models. To address this issue, researchers linked GNE-987’s hydroxyl group with six cysteines to an anti-CLL1 antibody, using an unstable carbonate linker to transform GNE-987 into a homogeneous DAC (Figure 5b). Data indicated that a single intravenous dose of 10 mg/kg DAC in subcutaneously transplanted HL-60 and EOL-1 AML mouse models could maintain sustained exposure in vivo and significantly inhibit tumor growth. Beyond BRD4, estrogen receptor-alpha (ERα), TGFβ receptor 2, and the chromatin regulator protein SMARCA2 (also known as BRM) have also been widely utilized as targets for DACs. However, DACs face the challenge that their payloads often possess high hydrophobicity, leading to overly hydrophobic overall structures. To ensure adequate cytotoxicity, a higher drug-to-antibody ratio (DAR) may be required, further exacerbating their hydrophobicity and adversely affecting the pharmacokinetics and toxicity profiles of these drugs. Thus, developing novel linker designs and improved degrader molecules to address these issues could significantly enhance the potential application of DACs in cancer therapy.
Dual-drug ADC
Dual-drug ADCs, by incorporating two different payloads, address tumor resistance and heterogeneity through the combined mechanisms of action of multiple cytotoxic drugs. This strategy may exhibit enhanced activity against heterogeneous tumor cell populations and resistant clones. As a single therapeutic entity, dual-drug ADCs could produce synergistic effects and overcome resistance in tumors that are non-responsive to treatment through a simplified administration regimen. Researchers initially hypothesized that delivering two payloads (MMAE and MMAF) simultaneously would enhance and synergize activity. Experiments showed that such dual-drug ADCs demonstrated potent activity in a mouse xenograft model of CD30-expressing anaplastic large cell lymphoma (ALCL) resistant to multiple drugs, resulting in the complete eradication of cancer cells in 3 out of 5 mice. In contrast, an ADC with a DAR of 8 carrying only MMAF showed lower activity, eradicating all cancer cells in only 1 out of 5 mice, while the equivalent ADC carrying only MMAE had no antitumor activity.
In a mouse xenograft model of Hodgkin’s lymphoma with heterogeneous CD30 expression, both the dual-drug ADC and the ADC carrying only MMAE completely inhibited tumor growth, likely due to MMAE-mediated bystander cell killing. Conversely, the equivalent ADC carrying only MMAF only moderately delayed tumor growth.
While dual-drug ADCs show potential advantages in overcoming tumor heterogeneity and resistance, caution must be taken regarding the potential for synergistic toxicity. To this end, researchers have developed various orthogonal conjugation strategies to produce dual-drug ADCs with high homogeneity and precise drug-to-antibody ratios. These strategies not only ensure effective integration of the two payloads but also optimize their combined action to maximize therapeutic effects and minimize adverse reactions.
More MMAE-linker complex product:
| Catalog | Product name | CAS NO | Inquiry |
| ADC-S-007 | Vc-MMAE | 646502-53-6 | Inquiry |
| ADC-S-008 | OSu-Glu-vc-PAB-MMAE | – | Inquiry |
| ADC-S-026 | MC-betaglucuronide-MMAE-2 | – | Inquiry |
| ADC-S-073 | mDPR-Val-Cit-PAB-MMAE | 1491152-26-1 | Inquiry |
Reference:
Tsuchikama, Kyoji, et al. “Exploring the next generation of antibody–drug conjugates.” Nature Reviews Clinical Oncology (2024): 1-21.