
Antibody-drug conjugates (ADCs) represent a highly promising approach to cancer treatment, capable of selectively delivering highly cytotoxic payloads to tumors. However, ADCs are currently facing a series of challenges, including resistance, tumor heterogeneity, and treatment-related adverse reactions. How can these challenges be addressed? On one hand, advancements in the components of ADCs (namely, the antibody, linker, and payload) are crucial for enhancing the efficacy and safety of the drugs. On the other hand, several emerging forms of ADCs, such as bispecific ADCs, conditionally activated prodrug conjugates (also known as probody-drug conjugates, PDCs), immune-stimulating antibody conjugates (ISACs), protein degrading ADCs, and dual-drug ADCs, offer their own advantages and are equipped to tackle various challenges:
- Bispecific and dual-drug ADCs possess the dual potential to combat resistance and tumor heterogeneity.
- Combining immune-stimulating and protein degrading ADCs with current treatment strategies may achieve multimodal therapy through several different mechanisms of action.
Recently, Nature Reviews Clinical Oncology published a review article that delves into the design of ADC molecules and how each component influences the drug’s properties. Furthermore, the article also highlights representative drugs of the aforementioned emerging ADC forms (bispecific ADCs, PDCs, ISACs, protein degrading ADCs, and dual-drug ADCs) that are in the preclinical/early clinical stages.
ADC molecular design
As shown in Figure 1a, ADCs are composed of a monoclonal antibody, payload, and linker, combining the specificity of the antibody with the high cytotoxicity of the payload. The principle of ADC design has evolved through the optimization of each structural component (antibody, payload, and linker).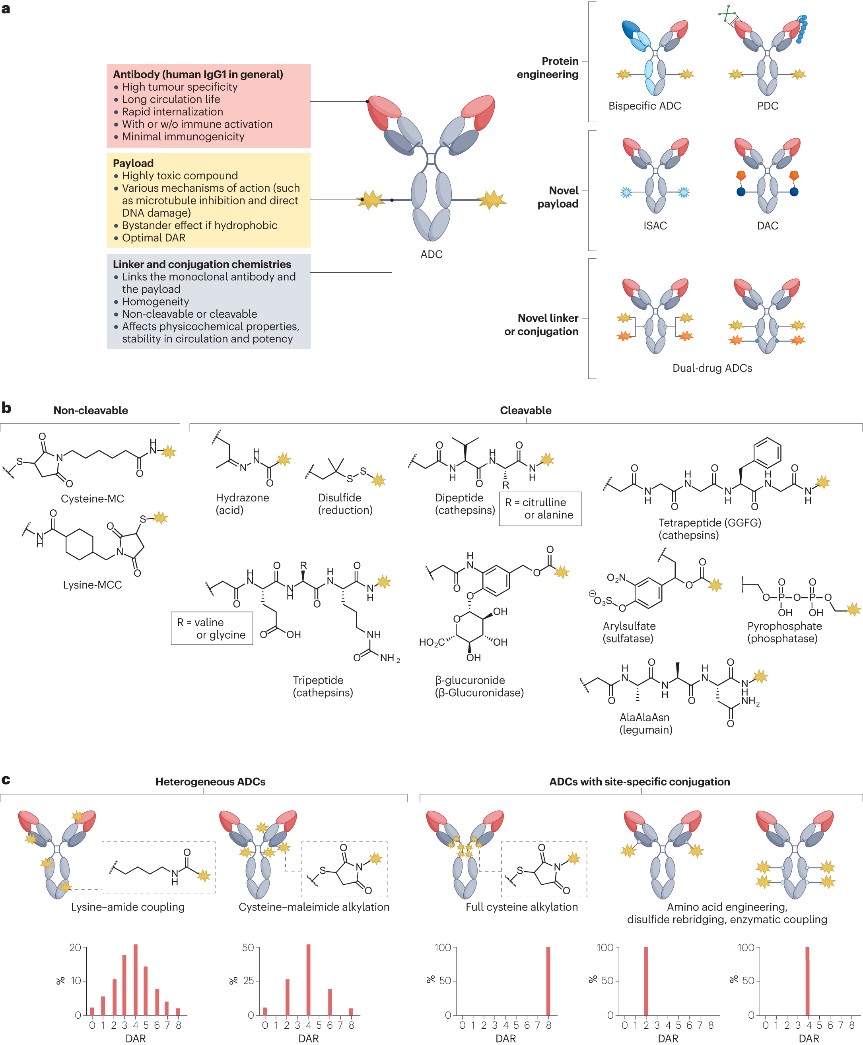
Figure 1. Composition, molecular properties, and new design of ADCs
- Selection of antibodies and targets
The antibodies used in ADCs are typically chosen to be either humanized or directly human immunoglobulin G (IgG). When designing ADCs, there is a preference for using IgG1 antibodies, as on the one hand, IgG1 is overall stable in the bloodstream and can synergize with innate immune cells (such as NK cells and macrophages); on the other hand, using human IgG1 also helps to reduce the overall immunogenicity of the ADC, thereby minimizing the risk of hypersensitivity reactions and the formation of anti-drug antibodies (ADAs). Most current ADC structures are linked to N-glycans to facilitate binding to FcγRs. However, glycosylation may lead to nonspecific uptake of ADCs by hepatocytes, leading to liver toxicity. In the future, non-glycosylated monoclonal antibodies may be used. Ideally, antibodies should recognize antigens that are specifically expressed on the surface of cancer cells and not expressed at all in healthy tissues. However, most ADC targets, such as HER2 and TROP2, are expressed to some extent in healthy tissues. Thus, both target-dependent and target-independent toxicities may still occur. Research is currently exploring antibodies that can specifically recognize structural variations in antigens present in tumors. Furthermore, the rate of ADC internalization and recycling also significantly impacts efficacy. Optimization of binding affinity is a crucial step towards maximizing the effectiveness of ADCs. Paradoxically, overly strong antigen binding can result in ADC molecules lingering on the surface of tumor cells, limiting the degree of tissue penetration. Therefore, different parameters must be considered in the antibody framework of an ADC to ensure optimal performance.
- Payload
The cytotoxic payloads of FDA-approved ADCs include antimitotics (MMAE, MMAF, and maytansinoid derivatives DM1 and DM4), DNA damaging agents (calicheamicins and pyrrolobenzodiazepine dimers), and topoisomerase I inhibitors (SN-38, DXd), among others. Other drugs currently under evaluation include microtubule inhibitors, DNA alkylating agents, topoisomerase II inhibitors, and RNA polymerase II inhibitors. Additionally, immunomodulators and protein degrader recruiters are gradually becoming promising new types of payloads. Most payload molecules possess a moderate-to-high level of hydrophobicity. Hydrophobic payloads can diffuse from the target-expressing tumor cells to adjacent cells with limited or no target expression, a phenomenon known as the bystander effect. Given that tumors with heterogeneity coexist with cells expressing the antigen and cells that do not, the bystander effect is crucial for the successful eradication of heterogeneous tumors. However, hydrophobic payloads can also negatively impact the efficacy of ADCs:
- Hydrophobic payloads can serve as good substrates for multidrug resistance proteins (such as MDR1, MRP1, and BCRP), reducing the efficacy of some ADCs against tumors that express these transport proteins.
- Hydrophobic ADCs tend to form aggregates, which may be rapidly cleared and could possess immunogenicity.
- Excessive hydrophobicity can promote liver uptake and cause hepatotoxicity.
Therefore, while ensuring the bystander effect, it is necessary to regulate the hydrophobicity of ADCs. One current strategy is to lower the drug-to-antibody ratio (DAR), but reducing DAR leads to decreased antitumor activity, so it is essential to finely tune the DAR for each payload category to balance efficacy and toxicity. Another solution is to incorporate hydrophilic masking groups, such as long polyethylene glycol (PEG) chains, which allows for the construction of high DAR ADCs while avoiding some of the undesirable effects of high hydrophobicity.
- Linker
As shown in Figure 1b, the linker plays a key role in keeping the cytotoxic payload attached to the antibody until it reaches the target. Linkers are classified into two categories: non-cleavable and cleavable. Non-cleavable linkers consist of stable chemical bonds that resist proteolytic degradation, providing excellent stability in the bloodstream. However, in ADCs with such linkers, the release of the payload requires the complete internalization and digestion of the antibody. In contrast, cleavable linkers are more widely used. These linkers can be degraded by tumor-associated factors (such as acidic and/or reductive conditions associated with most tumors or intracellular proteases). These linkers enable the effective release of the payload after internalization into cancer cells, thereby generating cytotoxicity, maximizing ADC efficacy, and facilitating the bystander effect. However, cleavable linkers carry the risk of premature payload release, leading to systemic toxicity and reduced efficiency of payload delivery. Therefore, striking a balance between stability and efficacy is crucial.
- Homogeneity
In addition to the structural components discussed above, achieving homogeneity in conjugation is also crucial. As shown in Figure 1c, traditional ADCs often use cysteine-maleimide alkylation and lysine-amide coupling for construction, but these random conjugation methods result in heterogeneous ADC mixtures with variations in payload attachment sites and drug-to-antibody ratio (DAR). The heterogeneity of ADCs typically leads to reduced efficiency of payload delivery, thus necessitating strict control over production to minimize such variations. There are now numerous conjugation technologies available for generating ADCs with specific DARs, such as interchain disulfide alkylation (used for T-DXd and sacituzumab govitecan), THIOMAB technology, conjugation involving non-natural amino acids, cysteine re-bridging, Fc affinity tags, and site-specific conjugation using various enzymes (such as engineered glycosyltransferases, transglutaminases, formylglycine-generating enzymes, and sortase).
Bispecific ADC
As previously mentioned, tumor heterogeneity and resistance can limit the antitumor activity of ADCs targeting a single antigen. To address this challenge, bispecific antibodies have emerged as a method capable of simultaneously binding two different target molecules and/or cells (Figure 2).
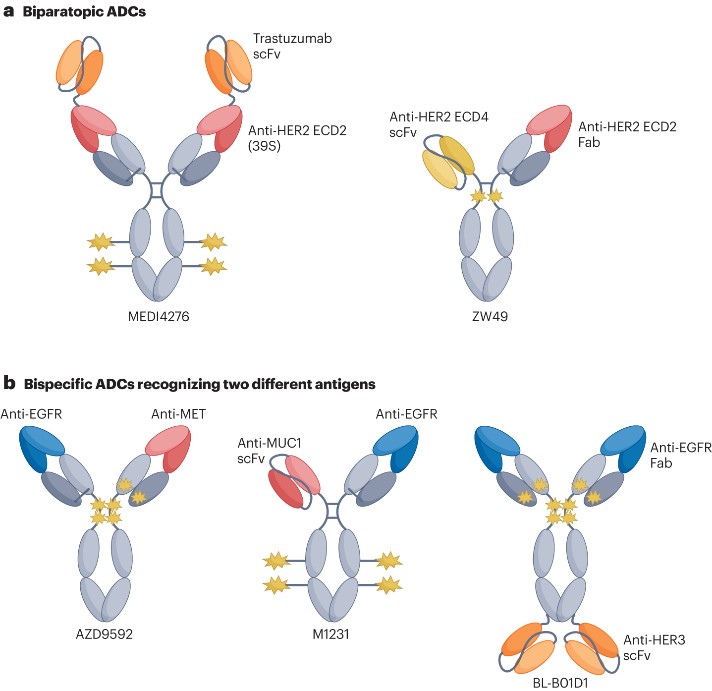
Figure 2. Representative bispecific ADCs
- Dual-epitope ADC
Previous studies have indicated that the use of two anti-HER2 antibodies binding different epitopes can lead to internalization, lysosomal transport, and downregulation of the target receptor. Therefore, researchers hypothesized that a multivalent bispecific ADC targeting two different epitopes on HER2 might improve binding affinity and enhance payload delivery. To test this hypothesis, researchers created the tetravalent HER2-targeting ADC MEDI4276 (Figure 2a) by fusing the single-chain variable fragment of trastuzumab with the N-terminus of another anti-HER2 IgG1 antibody, 39S. As expected, this homogenous bispecific ADC demonstrated faster internalization kinetics and lysosomal transport compared to trastuzumab or the parent 39S antibody. Despite showing high levels of activity in preclinical models, MEDI4276 did not exhibit a favorable efficacy-safety profile in clinical trials. In breast cancer patients, the objective response rate (ORR) was low (9.4%). The maximum tolerated dose (MTD) of MEDI4276 was determined to be 0.75 mg/kg every three weeks, but among the 12 patients receiving this dose, seven experienced one or more clinically significant and/or grade 3 or higher adverse events. Zanidatamab zovodotin (also known as ZW49) is another bispecific HER2 ADC (Figure 2a), which, unlike MEDI4276’s tetravalent binding, allows for bivalent binding to HER2 with its asymmetric structure. Despite differences in binding modality, ZW49 also induces HER2 receptor clustering and rapid internalization. A phase I dose-escalation study of ZW49 showed manageable safety and promising antitumor activity in heavily pre-treated patients.
- Bispecific ADCs targeting two different antigens
Bispecific ADCs, capable of simultaneously targeting two different antigens, offer multiple advantages:
- They can recognize and kill a broader range of tumor cells, including those from heterogeneous tumors.
- When targeting is optimal, bispecific ADCs can bind with higher tumor specificity, minimizing the risk of toxicity to healthy tissue.
- The involvement of multiple antigens and/or cells may produce synergistic effects.
As shown in Figure 2b, researchers developed the bispecific ADC AZD9592, which targets EGFR and MET (a molecule preferentially co-expressed with EGFR on the surface of tumor cells). This drug demonstrated promising monotherapy and combination therapy activity with osimertinib in EGFR mutant and wild-type NSCLC, as well as in head and neck squamous cell carcinoma PDX models. Moreover, AZD9592 was well tolerated in monkeys. These positive preclinical results have led to its Phase I trial (NCT05647122). M1231 is another bispecific ADC targeting MUC1 and EGFR, with SC209 as its payload. Preclinical data suggest that, compared to monospecific bivalent ADCs, it shows stronger internalization, lysosomal transport, and therapeutic activity in non-small cell lung cancer and esophageal squamous cell carcinoma (ESCC) PDX models. Additionally, BL-B01D1 is a tetravalent bispecific ADC targeting EGFR and HER3, with the camptothecin derivative ED04 as its payload, connected through whole cysteine coupling with a DAR of 8. Preclinical evaluation confirmed the compound’s antitumor activity in pancreatic or colorectal cancer mouse xenograft models, and it has entered Phase III clinical trials. However, the application of bispecific ADCs also faces potential challenges. For example, the expression ratio of the two target antigens can vary across different tumors and patients, potentially increasing the complexity of identifying and selecting potential treatment responders. Thus, careful evaluation of the target epitopes, binding modalities, and their potential biological properties is crucial for effective treatment with bispecific ADCs. This requires researchers to consider various biological and clinical factors comprehensively when developing bispecific ADCs to optimize treatment strategies.
Reference:
Tsuchikama, Kyoji, et al. “Exploring the next generation of antibody–drug conjugates.” Nature Reviews Clinical Oncology (2024): 1-21.