
The popularity of sulfhydryl groups in ADC preparation can be traced back to three factors: (a) thiols are the most nucleophilic amino acid side-chain functional group (much more nucleophilic than amines); (b) the relatively small number of cysteines (8 for IgG1 and IgG4) in the antibody suitable for conjugation; and (c) the fact that stability of the antibody is not critical, allowing for relatively controlled customization of the DAR; Interchain cysteines are present in the hinge region and connect the heavy chain (HC) and light chain (LC), appearing to favor “hiding” hydrophobic payloads after conjugation. It must be noted that a reduction step is always required prior to conjugation to release free thiols.
The vast majority of thiol-conjugated ADCs are maleimide-based (see Figure 1). Alkylation occurs between the primary interstrand cysteine (see Figures 1A and B) and the engineered cysteine (see Figure 1C). The exception is ADCs prepared by nucleophilic aromatic substitution. Other chemistries, such as disulfide exchange, are also being developed for some preclinical assets.
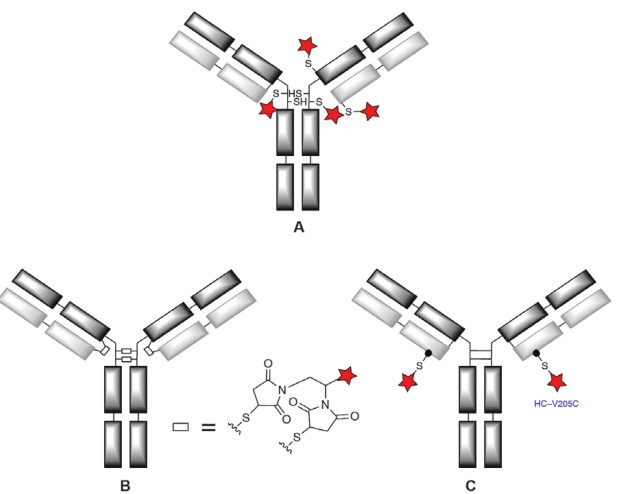
Figure 1. Groups based on maleimide chemically coupled ADC thiols1
Reaction with Maleimide
Maleimide is susceptible to alkylation by the nucleophilic Michael addition reaction of the thiol group to form a stable thioether bond (Figure 2). In the pH range of 6.5–7.5, the maleimide reaction is specific for thiols and reacts 1000 times faster with amines than at pH 7.0, so complete transformation can be achieved with only a small amount of excess reagent. The main drawback of the maleimide alkylation reaction is the reversibility of the Michael addition reaction, the rate of which has a lot to do with the pKa of the specific cysteine residue linked. This reverse Michael reaction may lead to the premature release of the linker payload in the bloodstream and may lead to adverse effects, which is also an important driver for the development of more stable maleimide variants.
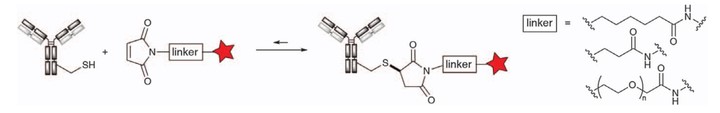
Figure 2. Alkylation of various maleimide reagents with cysteine1
As mentioned above, cysteines in monoclonal antibodies require a reduction step to release the thiol group prior to conjugation. As a result, a number of free thiol groups (up to 8 for IgG1 and IgG4) can be released by carefully optimizing the amount of reducing agent (typically TCEP or DTT) prior to the addition of maleimide-functionalized payloads. ADCs prepared by conjugation with cysteine include Adcetriss, Polivys, Padcevs, Enhertu, Blenrep, Trodelvys, Zynlontat, and others. Due to the lack of site specificity, these ADCs are typically produced as random mixtures with an average DAR of 2.3–4. However, there are two notable exceptions, Enhertu and Trodelvys, which, due to the relatively low toxicity of the camptothecin toxins carried, are produced by (near) quantitative conjugation of all interchain cysteines, with DARs of 7.7 and 7.6, respectively, and therefore can also be considered so-called site-directed coupling.
It is important to note that maleimide (mc) linkers are used in the vast majority of cases, although some ADCs use more electron-poor β-aminoethyl or ethylene glycol spacers. Some phenyl-substituted maleimides have also been reported to be more stable and are currently in preclinical development.
In 2008, it was reported that site-specific conjugation of the cytotoxic linker payload to monoclonal antibodies with specific cysteine may improve the therapeutic index of ADCs compared to randomized ADCs. Since then, a variety of site-specific ADCs have entered the clinic, such as SGNCD19B, CD123A, and CD352A (all HC-S239C mutations), RG7861/DSTA4637S (LC-V205C mutations), IMGN632 (S442C mutations), and BAT8003 (A114C mutations), and even bicysteine-based ADCs such as PF-06804103 (LC-K183C+). HC-K290C)。 In addition, two complementary technologies have been developed, namely cysteine insertion (e.g., MEDI2228 (HC-i239C)) and fusion of HC-terminal peptide tags (e.g., ALT-P7 (C-terminal ACGHAACGHA)). Unfortunately, the use of cysteine-engineered antibodies does not guarantee clinical success, as some of these assets have been abandoned (e.g., BAT8003 and all three Seagen ADCs mentioned above).
The inverse Michael reaction can be inhibited by hydrolyzing the original thioether-succinimide adduct to provide a maleamide acid derivative. This hydrolysis can be achieved by prolonged treatment of ADCs at pH 9. Such conditions may apparently result in additional post-translational modifications of the antibody, such as deamidation or pyroglutamic acid formation. In response to this problem, Seagen has developed an effective solution by adding aminomethyl maleimide, which is based on 2,3-diaminopropionic acid (DPR technology), to the α position of the N-alkyl chain (see figure below). The amino group induces spontaneous autohydrolysis of the succinimide conjugate, thereby blocking the inverse Michael reaction. The ADC product SGN-CD48A (now abandoned) was the first to feature DPR technology, as was SGN-CD228A.
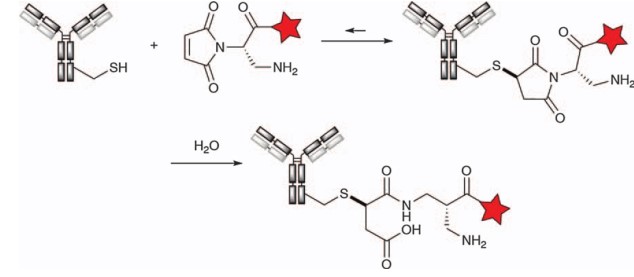
Figure 3. Cysteine is alkylated with a diaminopropionic acid (DPR)-based aminomethyl maleimide reagent and then autohydrolyzed to obtain maleic acid conjugates.1
Our drug-linker complex products:
| Cat.No. | Product name | CAS NO | Price |
| ADC-S-024 | DBM-MMAF | – | Inquiry |
| ADC-S-025 | MC-betaglucuronide-MMAE-1 | – | Inquiry |
| ADC-S-026 | MC-betaglucuronide-MMAE-2 | – | Inquiry |
| ADC-S-027 | SGD-1910 (Mc-Val-Ala-PBD) | 1342820-51-2 | Inquiry |
| ADC-S-028 | MC-PBD | – | Inquiry |
Other Cysteine Alkylation Reagents
For example, the high reactivity of α-haloacetamide reagents is well known, and in the case of 2-iodoacetamide, it is an indisputable choice for blocking free cysteine in proteins or protein mixtures (e.g., cell lysates). Importantly, the resulting thioether bond is completely irreversible and therefore superior to thiol-maleimide ether in terms of stability. In fact, Alley et al. reported in 2008 that the use of a-bromoacetamide hexanoyl linkers could provide a thioether ADC with excellent plasma stability, with no measurable systemic drug release within 2 weeks of mouse dosing and a 25% increase in intratumoral drug exposure within 7 days. However, despite the differences in stability between bromoacetamide and maleimide-based ADCs, there were no significant differences in their potency, activity, or toxicity properties. The reason for this surprising lack of distinction is likely to be that for this particular maleimide-based ADC, the half-life of the ADC’s linker and the half-life of the ADC clearance are similar. Since ADC concentrations decrease over time, prolonging the junction half-life beyond the pharmacokinetic half-life is unlikely to have a significant effect on drug exposure.
Based on the discovery that methylsulfonylbenzothiazole (MSBT) is a selective thiol blocker, Barbas et al. developed a series of aromatic methylsulfonone reagents for the alkylation of thiol-containing proteins, mainly oxazinazole and benzothiazole. Similarly, methylsulfonate reacts with thiols through a nucleophilic aromatic substitution mechanism (see figure below), a concept that is applied in SKB264/BT001035.
Researchers at Glythera have reported highly stable thioether ADCs obtained by incubating free cysteine thiols with 4-vinylpyridine, a technique known as PermaLinks. The resulting ADC is significantly stable and is not easily degraded.
The last technology worth mentioning is the phosphoamide coupling technology developed at the University of Berlin, which is still in its early stages but is currently being commercially used by Tubulis. Studies have shown that aryl phosphonate with electron-deficient triple bonds has excellent cysteine-selective reactivity, and the resulting (Z)-vinyl sulfide has excellent stability compared to traditional maleimide linkages.
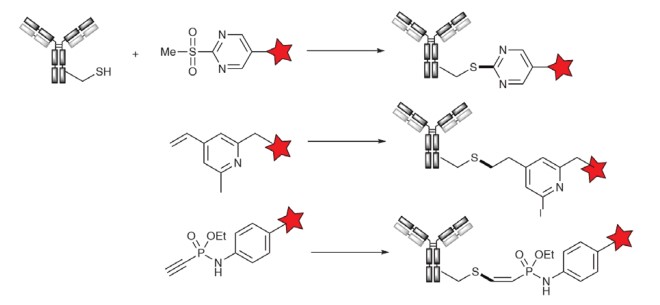
Figure 4. Alternative techniques for cysteine alkylation1
Alkylation/Crosslinking with Bismaleimide or Bismaleamide Acid
The ability to alkylate maleimide has also been applied to various cross-linked bismaleimide reagents. Each reagent captures two free cysteine thiol groups, resulting in the formation of DAR4 ADCs after the complete reduction of all eight interchain disulfide bonds. It is important to note that although mixtures are generated due to interchain cross-linking between thiol groups (e.g., from HC-226C to HC-229C), the resulting ADCs have a narrow DAR distribution. NewBio’s NBT828 uses a bismaleimide crosslinker. Bisuccinimide conjugates may also be hydrolyzed to carbamate derivatives upon the nucleophilic addition of a thiol group, providing a conjugate that cannot undergo reverse Michael degradation.
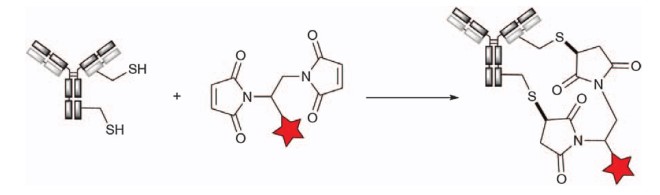
Figure 5. Bismaleimide reagent cross-linked cysteine1
Researchers from AstraZeneca reported rebridging cysteine-engineered antibodies with bismaleimide-modified payloads (PBDs) to obtain DAR1 ADCs.
THIObridget Conjugation
A novel coupling reagent originally reported by Del Rosario et al. and Wilbur et al., taking advantage of the high nucleophilicity and susceptibility to Michael addition reactions of free thiols, was further developed by Poly-Therics (now known as Abzena, with THIObridget technology) for ADCs. This rebridging technique enhances the stability of the reducing antibody by using the thiol formed by religating the dithiol reaction group, more effectively maintaining the conformational integrity of the final conjugate. Close-to-the-molecular-scale dithiol reactive groups are well suited for this purpose, as they can be seamlessly integrated into the protein structure without disrupting subunit interactions. The a-toluenesulfonyl methyl-a,b-unsaturated ketone reagent was applied to the reducing antibody, which was generated in situ from a precursor of bis-a,a-toluenesulfonyl methyl ketone, triggering a series of reactions including Michael addition, b-elimination toluenesulfonic acid, and subsequent Michael addition to produce a stable conjugated product.
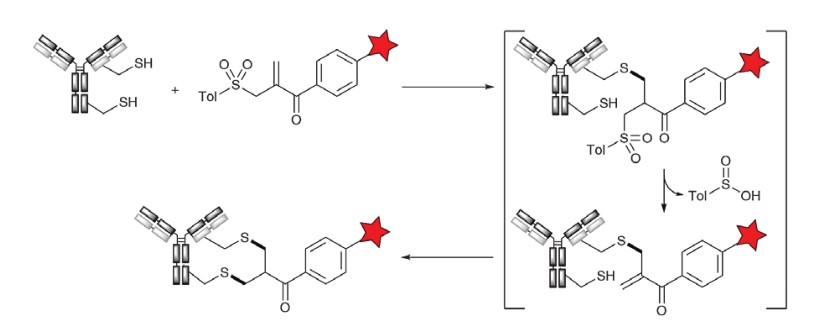
Figure 6. THIObridget technology1
Alkylation/Conjugation by Bisbromomethyl Aromatic Reagents
Concortis (now Sorrento) developed a third coupling method for DAR4 ADCs, called C-lockt technology. The C-lockt technique combines the nucleophilic nature of thiols with the propensity of the benzyl bromide population to undergo nucleophilic SN2 substitution reactions. Thus, by reducing the interchain disulfide bonds, which then react with a bisbromomethyl aromatic reagent (e.g., bisbromomethylquinoxaline), an irreversible cross-linking reaction occurs to form a thioether. Interestingly, it was reported that the clinical ADC (CD38-ADC) prepared using the C-lockt technique was not DAR4 but DAR3 (averaged).
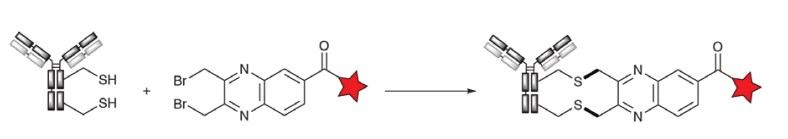
Figure 7. Cysteine crosslinking by diaalkylation of bisbromomethylquinoxaline1
Use Dibromopyridine Dione for Conjugation
Inspired by the use of dibromomaleimide groups to re-bridging reduced disulfide bonds, Chudasama et al. developed a rebridging coupling technique based on the pyridine dione (PD) core structure. Unlike maleimide derivatives, pyridine dione rings are more stable and do not open the rings by hydrolysis, resulting in a more homogeneous binder. The authors prepared monobromo-PD (MBPD) and dibromo-PD (DBPD) derivatives and demonstrated that both react specifically with thiol groups at pH 8.0, even in the presence of amines. DBPD reactive groups have been used to develop a range of crosslinking and modification reagents that incorporate other reaction sites, such as alkynes for click chemistry and even derivatives containing reducing agents, for one-step antibody reduction and conjugation. The use of DBPD thiols to rebridge chemistry with antibody binding has been applied to create highly fixed-point ADCs, such as trastuzumab for DAR4 MMAE.
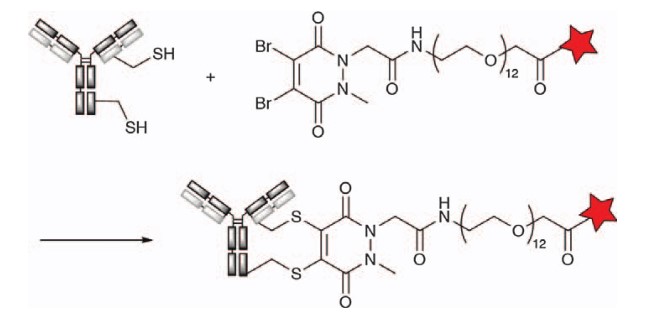
Figure 8. Cross-linking of antibody thiol groups with dibromoazadione derivatives1
Disulfide bond coupling
Disulfide bonds have been used since the early days of ADCs, such as in MLN2704, CMD-401, BIWI-1, and IMGN242. A linker design containing a disulfide bond is located somewhere between the cytotoxic payload and the antibody and can facilitate a reversible linkage that can be cleaved by glutathione reduction within tumor cells to release the toxin. However, the reality is that premature severing of circulating disulfide bonds often occurs due to the presence of free cysteine, which results in the release of the payload before the tumor cells enter. The addition of a protective group, such as a methyl or dimethyl group, to the carbon adjacent to the disulfide bond has been used to increase stability in circulation by creating steric hindrance around the disulfide bond, but in many cases, it also reduces the rate of toxin release from tumor cells. In addition, even though the increased steric hindrance improves the stability of disulfide bonds, these ADCs are not as effective as conjugating drugs to engineered cysteine residues at optimal locations in the antibody sequence.
For the first time, Pillow et al. directly conjugated a small molecule drug to an engineered cysteine antibody at different locations to generate a sterically unhindered disulfide bond-Mertansine ADC. The results were compared with ADCs with different sterically hindered disulfide bonds conjugated to lysine residues. The authors identified the engineered cysteine located at LC-K149C as the most stable binding site for drug attachments containing disulfide bonds. ADCs created using the LC-K149C locus show excellent circulatory stability while allowing potent glutathione (GSH)-mediated cleavage within tumor cells and thus have the highest potency against mouse human non-Hodgkin lymphoma xenografts of all mutants evaluated.
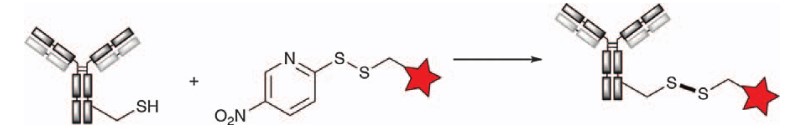
Figure 9. Disulfide bonds are produced by engineering cysteines to prepare antibody conjugates1
Reference:
- van Delft, Floris, and John M. Lambert, eds. Chemical Linkers in Antibody-Drug Conjugates (ADCs). Vol. 81. Royal Society of Chemistry, 2021.