
Introduction of the Camptothecin Derivative Payload
Camptothecin was first isolated from camptothecin stems in 1966 by Wall et al., mainly in the form of topological isomerase (topoI). In order to inhibit the synthesis of DNA as a target to exert an anti-cancer effect, its derivatives have been widely used in the treatment of related cancers in clinical practice, including irinotecan and topotecan. The success of DS-8201 has pushed the research of camptothecin to a climax. According to statistics, there have been 72 ADC drugs carrying camptothecin derivatives into the clinic. These 72 ADCs target a variety of different targets, including B7-H3, HER3, TROP2, Nectin4, FRα, Cldn18.2, MUC18, CDH6, HER2, cMET, etc., and in addition, these ADCs also use up to 21 different linkers and 15 different payloads.
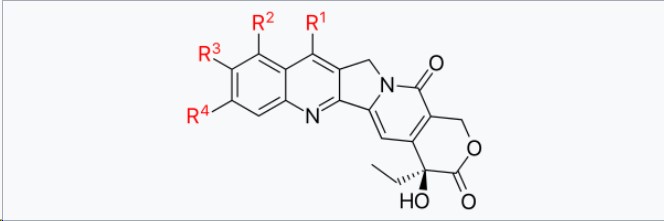
Figure 1. A Schematic View of the Camptothecin Analog
Considerations for the Modification of the Structure of Camptothecin
Let’s take a look at how Zymeworks’ Payload was designed and screened, starting with determining the location of the modification based on the current research on camptothecin. In order to design a new camptothecin analog, the researchers analyzed more than 60 camptothecin structure-activity relationship (SAR) data. The camptothecin structure that is essential for activity includes the 20(S)-hydroxyl and lactone functional groups of the E ring, the pyridone D ring, and maintaining the planarity of the ABCDE ring system. The C-7 and C-9 positions can accommodate a variety of substituents, including additional fusion rings as seen in exatecan and its derivatives, while substitutions in the C-12 to C-14 regions significantly reduce activity. C-10 and C-11 can only accommodate small substituents, and 11-fluoroptothecin shows higher activity, which makes it possible to explore a wider variety of substituents at the C-7, C-9, and C-10 positions.
Our Ready-To-Use Camptothecin Analog Products
| Cat.No. | Product Name | Price |
| ADC-W-372 | Anti-CD70 (clone h1F6)-β-glucuronide-Camptothecin Analog ADC | Inquiry |
| ADC-W-349 | Anti-TNFRSF8 (CA10)-β-glucuronide-Camptothecin Analog ADC | Inquiry |
| ADC-W-2563 | Anti-MSLN (Anetumab)-MC-Vc-PAB-SN38 ADC | Inquiry |
| ADC-W-1986 | Anti-Phosphatidylserine (Sevirumab)-MC-Vc-PAB-SN38 ADC | Inquiry |
| ADC-W-2154 | Anti-IgE Fc (Talizumab)-MC-Vc-PAB-SN38 ADC | Inquiry |
Preliminary Screening of Candidates for Payload
Based on the above study, Zymeworks focused on the modification of the C-7 (R1) and C-10 (R3) positions of the camptothecin backbone, while keeping the 11-fluorosubstituent unchanged and C-9 unsubstituted. His research focuses on analogs with hydrophilic functional groups (e.g., amines, carbonates, urea, sulfonamides) with a linkable hydroxyl group at the C-7 position and a methyl or methoxy group at the C-10 position. In addition, the investigators also sought to evaluate camptothecin analogs with an amino group at the C-10 position, providing an alternative for linker connection. Ultimately, a library of approximately 100 paclitaxel small molecules was synthesized and evaluated in vitro for cytotoxicity in a range of HER2-expressing cell lines, including SK-BR-3, Calu-3, SK-OV-3, and ZR-75-1.
Structure-activity relationship analysis of these synthesized small molecules revealed that most compounds with methyl groups at the C-10 position had a pIC50 ≥ 8.0 (i.e., IC50 < 10 nM) with large variability in their hydrophobic profiles (cLogD range of -1 to 3). analogs with an amine group at the C-10 position are comparatively more effective and more hydrophilic. In contrast, camptothecin with a C-10 methoxy substituent was less active and had no significant improvement in hydrophobicity.
Payload-linker Combination Filtering
From this library, seven derivatives were selected for further study with methyl or amine groups at the C-10 position and covering a wide range of activity, hydrophilicity, and structural diversity at the C-7 position. Subsequently, these seven different derivatives were studied with different linkers and different positions, and the linker is a cleavable tetrapeptide sequence GGFG, which is combined with maleimide containing PEG or maleimide-triethylene glycol (MT) propionyl group. In addition, DL7 and DL8 were prepared for direct comparison with ADCs carrying clinically validated DXd vectors.
Comprehensive Screening of Druggability
This was followed by a comprehensive evaluation of the different drug candidates, including hydrophilicity, in vitro target-dependent efficacy, stability (blood stability, stability of PH7 buffer species), bystander effect, efficacy in animal models, including 3D tumor model efficacy, ADME/DMPK properties, tolerability in non-human primates, etc., and finally determining the candidate Payload ZD06519 (FD1).
Summary
The success of DS-8201 completely detonated the research on ADCs, and at the same time, it also put the camptothecin derivative DXd in the spotlight and attracted countless imitations from later generations. Payload’s modification is a systematic project, and any small changes will not only affect the compound itself but may also affect the final efficacy. This article introduces in detail the process of ZD06519 (FD1) modification screening, including the consideration of modification location, the consideration of substitution group, the consideration of physical and chemical properties, the consideration of metabolism and pharmacodynamics, etc., which not only provides a reference for the modification of camptothecin derivatives, but also provides a direction for the development of other payloads.