
The PD-1/PD-L1 pathway, a critical immune checkpoint in the human body, is influenced by numerous regulatory factors, ranging from genetic mutations and variations to gut microbiota, as well as various factors inside and outside cells, including epigenetic modifications.
In a recent study published in Science Advances, Professor Guihua Wang and his team from Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, revealed that the methylation level of PD-L1 significantly affects the interaction between PD-1 and PD-L1. This interaction, in turn, impacts the anti-tumor immune response and the effectiveness of immunotherapy.
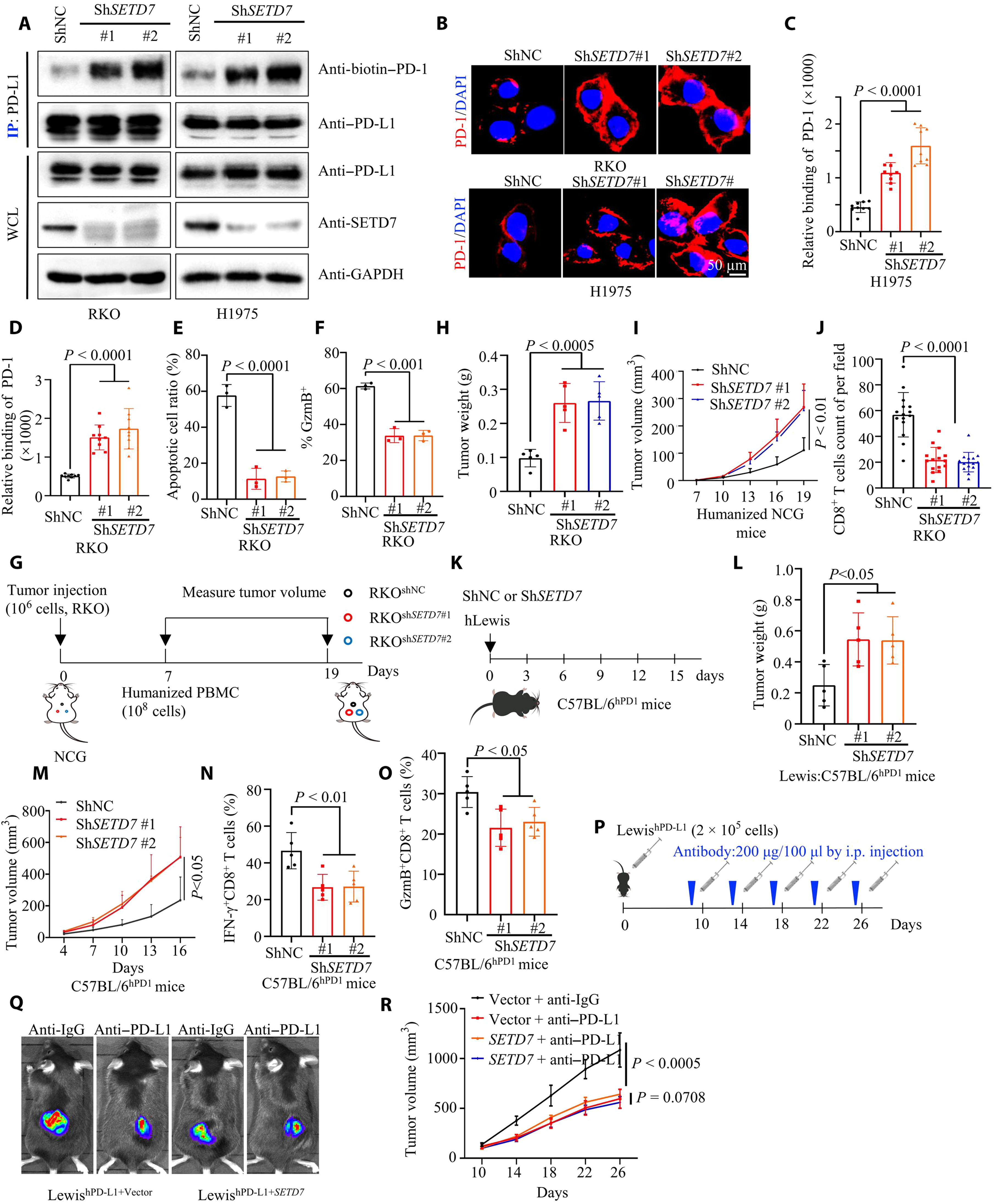
Fig.1 PD-L1 methylation restricts PD-L1/PD-1 interactions to control cancer immune surveillance (Huang, 2023)
The study identified lysine 162 (K162) as a key methylation site for PD-L1. SETD7 methylates this site, while LSD2 demethylates it. High methylation at this site is a key mechanism leading to resistance to PD-L1 inhibitors. Consequently, the methylation level at K162, compared to the overall PD-L1 level, can serve as a biomarker for predicting the efficacy of immune therapy.
Previous studies have shown that post-translational modifications such as ubiquitination, glycosylation, phosphorylation, palmitoylation, and acetylation, play crucial roles in regulating the PD-1/PD-L1 pathway. The impact of methylation on this pathway has been hypothesized, but specific mechanisms require further clarification to identify specific target sites and predictive indicators.
To investigate this, the research team treated human renal epithelial HEK293T cells transfected with PD-L1 using a methylation inhibitor called Dznep. After immunoprecipitation, mass spectrometry analysis is performed to identify lysine/arginine methylation sites on the PD-L1 protein that might affect the interaction between PD-1 and PD-L1. The K162 site was selected among five other sites. Subsequent experiments revealed that the absence of methylation specifically at the K162 site of PD-L1, without affecting the expression level of PD-L1, significantly enhanced the binding ability of PD-1 and PD-L1 (stronger than wild-type PD-L1) and altered their interaction state.
When the K162 methylation site of PD-L1 was “knocked out” in T cells, the cytotoxicity of CD8+ T cells significantly decreased. This decrease was indicated by reduced expression of granzyme B and interferon-γ. Similarly, in immunodeficient mice treated with the same approach, the growth rate of transplanted tumors increased significantly. This increase was primarily due to a decrease in the number of CD8+ T cells within the tumors and a reduction in immune surveillance function.
Combining the conclusions from these two sets of experiments, it can be concluded that a decrease in the methylation level of the PD-L1 K162 site significantly weakens the cancer immune surveillance mediated by the PD-1/PD-L1 pathway. This weakening leads to tumor occurrence and development. The next step is to identify key enzymes that regulate methylation.
The study demonstrated that SETD7, a histone lysine N-methyltransferase, is the only enzyme capable of upregulating methylation at the PD-L1 K162 site. Deletion or mutation of the SETD7 gene, as well as abnormal interleukin-6 (IL-6) signaling, all affect the methylation of PD-L1 at K162. This influence affects the interaction between PD-1 and PD-L1, ultimately weakening the anti-tumor immune response. However, overexpression of SETD7 triggers resistance of tumors to PD-L1 inhibitors.
Conversely, lysine-specific demethylase 2 (LSD2), which can downregulate the methylation level of PD-L1 K162, acts in the opposite manner. It can decrease the methylation level of PD-L1 K162. LSD2 has a similar impact on the anti-tumor immune response in mice as the knockout of PD-L1 K162 methylation. Additionally, overexpression of LSD2 can enhance tumor sensitivity to PD-L1 inhibitors.
Based on these experimental results, the research team suggests that the methylation level of PD-L1 K162 can be used to predict the clinical efficacy of PD-1/PD-L1 inhibitors. They validated their findings using the TIMER/GSCA tumor immune database.
The analysis showed that the methylation level of PD-L1 K162, or the ratio of PD-L1 K162 methylation to the overall PD-L1 expression level (MPR), positively correlates with the infiltration quantity and cytotoxicity of CD8+ T cells within tumors. Patients who respond to PD-L1 inhibitors exhibit high expression of LSD2 and PD-L1, as well as low expression of SETD7. They also have a lower methylation level at the PD-L1 K162 site.
Using an MPR < 0.65 as a cutoff value, the specificity and sensitivity for predicting the response to PD-1 inhibitors in the treatment of non-small cell lung cancer (NSCLC) were 77.27% and 93.33%, respectively. An MPR < 0.65 was also associated with longer overall survival (OS) in patients, indicating that a lower methylation level at the PD-L1 K162 site is more favorable for immune therapy. This finding aligns with the experimental conclusions.
The researchers point out that patients with an MPR > 0.65, indicating a high methylation level at the PD-L1 K162 site, may have diminished cytotoxicity of CD8+ T cells within tumors after the binding of PD-1 and PD-L1. Therefore, PD-1/PD-L1 inhibitors may not be suitable for these patients, and alternative types of immunotherapeutic drugs, chemotherapy, radiotherapy, or other methods should be considered as priorities.
In summary, this study confirms that the methylation status of PD-L1 has a significant impact on anti-tumor immune responses and the efficacy of PD-1/PD-L1 inhibitors. Monitoring the methylation level of PD-L1 at K162 can serve as a novel strategy for predicting the efficacy of immune therapy and guiding precise clinical medication.
Reference
1. Huang, Changsheng, et al. “PD-L1 methylation restricts PD-L1/PD-1 interactions to control cancer immune surveillance.” Science Advances 9.21 (2023): eade4186.