This article introduces a research work published in nature structural & molecular biology. The author explores the latest progress and limitations of deep learning methods in predicting the structure of human protein interactions, explores the potential regulatory mechanisms in interface residues, introduces the use of predicted binary complexes to construct multimers, and advances our understanding of human cell biology.
Introduction
Structural characterization of protein complexes is a key step in understanding protein functional mechanisms, studying the effects of mutations, and studying cellular regulatory processes. Recently, neural network-based methods have gained the ability to accurately predict individual proteins and protein complexes, but their utility for large-scale human complex structure prediction has not been validated. Here, the authors assessed the possibilities and limitations of applying AlphaFold2 to model the structure of large-scale human protein interactions. The authors predicted 65,484 complex structures derived from different experimental methods, of which 3137 predicted structures had high confidence, and they were enriched in the predictions supported by the combination of experimental methods, confirming the possibility of evaluating prediction models according to confidence. By studying pathogenic mutations and phosphorylation of interface residues, the authors demonstrated the value of structurally resolving the interactome. Finally, the authors also presented a case of building multimers by using binary complexes.
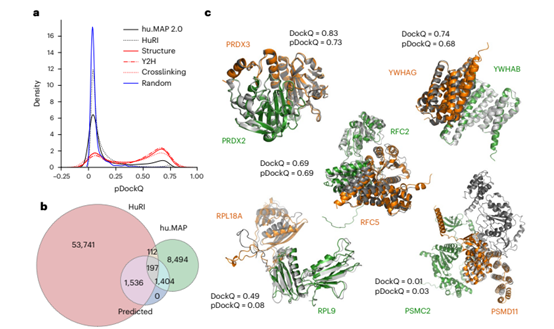
Figure 1. Application of AlphaFold2 complex prediction on a large-scale human PPIs dataset
Result
Structural Prediction of Human Protein Interactions
The authors used the AlphaFold2-based FoldDock pipeline to predict 65,484 experimentally determined human protein interaction structures derived from the HuRI and hu.MAP v.2.0 databases. As in the FoldDock pipeline, the authors combined the size of the interface with the plDDT score into pDockQ to predict the DockQ score of the complex, which ranked the models according to their confidence. In Figure 1, the authors showed the distribution of pDockQ scores for protein interactions in different scenarios. The results showed that proteins with known interactions tend to have higher pDockQ scores than random ensembles and predict higher confidence for the hu.MAP dataset than for the HuRI dataset. This indicated that models with high confidence were enriched in protein interaction regions with high affinity and direct interactions. In addition, the authors showed examples of predicted structures aligned to experimental models or cognate models, illustrating how predictions and confidence scores related to observed alignments. Taken together, the experimental results demonstrated that AlphaFold2 can predict the structures of directly interacting protein pairs in large complexes.
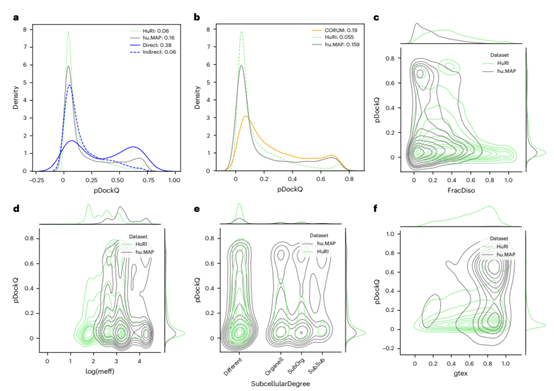
Figure 2. Protein and interaction features affecting prediction confidence: analysis of different datasets
Features that Affect Prediction Confidence
As shown in Figure 1, compared with the protein pairs in the HuRI and hu.MAP databases, the protein pairs appearing in the PDB library were more enriched in the high-scoring model part. To better understand this discrepancy, the authors first investigated an additional dataset consisting of large heterogeneous protein complexes in which protein pairs could be classified according to whether they had direct interactions. The prediction results showed that the difference in pDockQ scores between direct versus indirect interacting pairs was significant, suggesting that direct interacting pairs were often predicted even when they were part of large complexes. In order to explore the reason for the difference between the prediction confidence of hu.MAP and HuRI database, the author introduced the CORUM database as a control. Experimental results showed that the proportion of high-confidence predictions was similar between different protein complex databases. The HuRI database was an exception, probably because many protein interactions in the HuRI database were transient and AlphaFold2 happened to be unable to predict effectively.
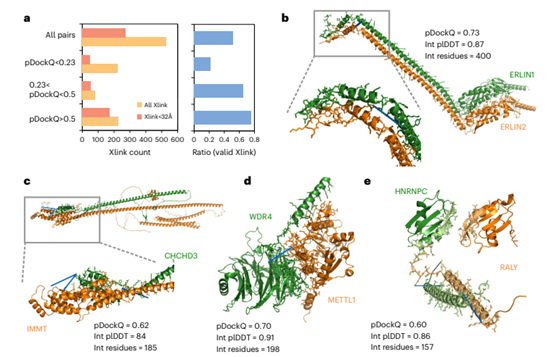
Figure 3. Chemical cross-linking support for predictive complex models
Validation of Predicted Complex Structures on Chemical Crosslinks
Chemical cross-linking mass spectrometry is a method that can be used to identify nearby reactive residues, and the identification of such residues in a pair of proteins can aid in the definition of possible protein interfaces. To determine whether the predicted complex structure satisfies this orthogonal spatial constraint, the authors acquired a collection of residue-pair crosslinks for 528 protein pairs with predicted models. Of these, 51% of the models had one or more crosslinks at distances below the expected maximum distance. When the pDockQ score was upregulated to constrain the confidence of the predictive model to a higher level, the proportion of complexes with acceptable crosslinking also increased. The proportion reached 75% when the pDockQ score was greater than 0.5, which was consistent with the benchmark results. The authors then provided multiple cases in Figure 3 to illustrate the effectiveness of chemical crosslinking validation.
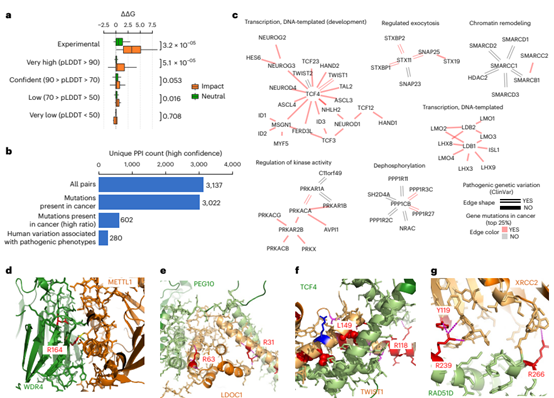
Figure 4. Disease mutations of protein complex interface residues
Disease-associated Missense Mutations Located at Complex Interfaces
To determine the usefulness of the predicted structure, the authors compiled a set of mutations located at interface residues that had been experimentally tested for their effect on the corresponding interactions. The authors used FoldX on these mutations to predict mutational changes in binding affinity and observed that interaction-disrupting mutations strongly affected binding stability. Further experiments confirmed that only very accurate models are valid when using the FoldX force field to estimate the effect of mutations on binding affinity. The authors then mapped human disease and cancer mutations to interface residues defined by high-confidence protein complex prediction sets. They found a strong enrichment for disease-causing versus benign mutations at interface residues relative to the rest of the protein. Finally, in Figure 4, the authors exemplified protein network clusters with mutations in interface residues and interface residues with disease mutations.
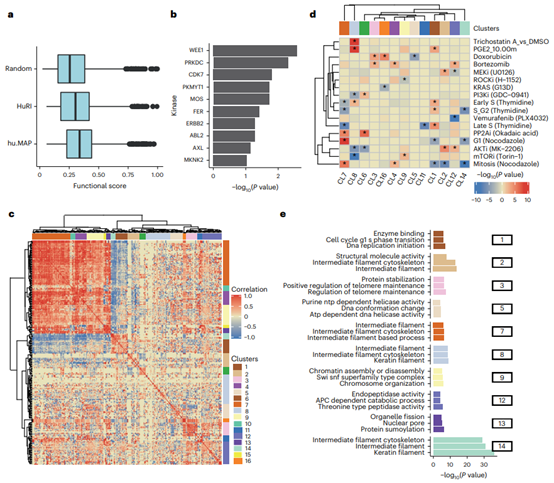
Figure 5. Co-regulation of interface residue phosphorylation sites
Phosphorylation Regulation of Protein Complex Interfaces
Mapping the positions of phosphorylation sites to protein interfaces can provide mechanistic hypotheses for their roles in controlling protein interactions. Using recent descriptions of human phosphoproteins, the authors identified 4145 unique phosphate groups at interface residues in a high-confidence model. Phosphorylation sites at interfaces were also found to be of higher functional importance than random sites, and some interfaces may be coordinated by specific kinases and conditions. Afterwards, the authors delved into and demonstrated potential co-regulatory interfaces with examples. While not all phosphates likely modulated binding affinity, this analysis provided the hypothesis for coordinated regulation of multiple proteins by modulating interactions following specific perturbations.
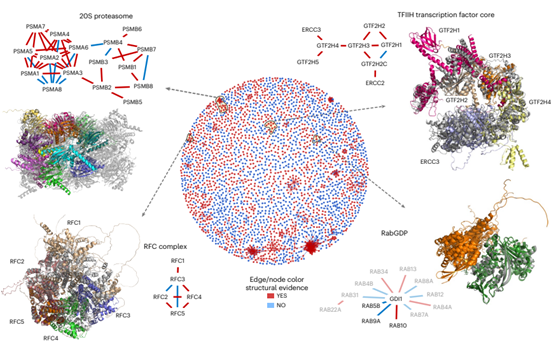
Figure 6. High-order combinatorial prediction of protein complexes
Higher-order Combinations of Binary Protein Interactions
Proteins can both interact with multiple proteins simultaneously, as part of larger protein complexes, and be separated in time and space. This is also reflected in the authors’ proposed network of structural features, whereby proteins can be discovered in groups, as shown in the global network view of protein interactions. Due to computational resource constraints, the authors only tested the process of iterative structure alignment on a small set of complexes covered by the network, and analyzed the potential and limitations of the process by aligning experimentally determined structures with predicted models. Encouraged by test examples, the authors designed an automated process to generate larger complexes through iterative alignments, and further analyzed some of the factors limiting the success of this process after confirming its feasibility.
Conclusion
The author analyzed the feasibility of using AlphaFold2 for protein complex prediction through a series of experiments, and noticed that the source of data has an important impact on the prediction effect. The analysis showed that combining protein interactions supported by affinity-based, co-segregation techniques, and complementarity methods can lead to higher confidence models. The authors proposed that the evaluation metrics derived from the models can be used to rank high-confidence models to aid in the study of large-scale PPIs and stable complexes, and that cross-linked mass spectrometry experimental data can provide an ideal resource for further validation of these ideas. In addition, in-depth analysis of missense mutations and phosphorylation of interface residues was also conducive to further understanding molecular mechanisms. At the end of the article, the author put forward the idea of building a larger complex structure model starting from the binary complex, and analyzed its potential and limitations, which provided a broad idea for the construction of multimers.
References
Burke, D.F., Bryant, P., Barrio-Hernandez, I. et al. Towards a Structurally Resolved Human Protein Interaction Network. Nat Struct Mol Biol (2023). https://doi.org/10.1038/s41594-022-00910-8