
Antibody-drug conjugates (ADCs) comprise an antibody, a linker, and a cytotoxic active payload. Compared to traditional chemotherapy, ADCs that target tumors are expected to mitigate systemic toxic effects, provide a wider therapeutic window, and have a higher therapeutic index. Over the past decade, ADCs have progressively matured in the treatment of solid tumors and hematological malignancies, becoming a revolutionary field in anti-tumor drug research.
While the development of ADCs has achieved significant success, the occurrence of unexpected toxicity and resistance presents substantial challenges. Several factors have led to a common clinical dilemma, particularly in solid tumors. These factors include:
(1) The heterogeneity of solid tumors restricts the clinical efficacy of targeting a single antigen.
(2) Therapy-induced antigen down-regulation, epitope mutation, and the activation of bypass pathways lead to significant ADC resistance.
(3) Off-target toxicity results from antigen expression in normal tissues, linker instability, and other variables.
(4) The resistance of target antigens to internalization hampers adequate therapeutic effect.
Therefore, the optimization of the antibody, linker, and active payload is a challenge for next-generation ADCs.
A prospective way to address these clinical challenges consists of the conjugation of bispecific antibodies (BsAbs) with linker-cytotoxic payload complexes, which has led to the creation of bispecific antibody-drug conjugates (BsADCs). Compared to traditional ADCs, BsADCs have a unique dual epitope/target binding mode that can not only bind with co-expressed antigens in solid tumors to enhance selectivity, but also significantly improve internalization. These unique advantages make BsADCs a significant force in the next generation of ADCs. Currently, more than 10 different BsADCs are being tested in clinical trials. The design of BsADCs is not just “1+1=2”, but it involves a comprehensive harmonizing and optimizing of BsAbs, linkers, and cytotoxic payloads where the change in binding modes influences the overall therapeutic effect.
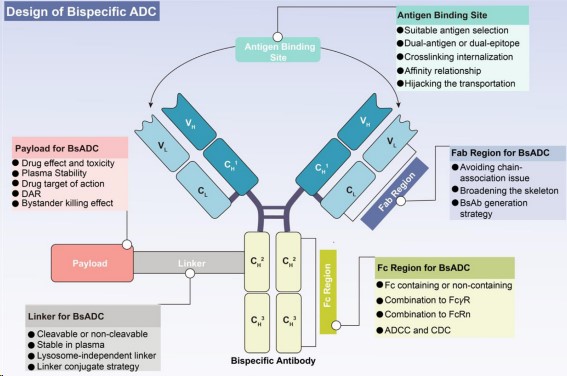
Fig.1. The concept and design considerations of BsADCs (Gu Y,2024)
Design of BsADCs
The known targets of BsADCs are mainly concentrated on HER2, EGFR, and c-MET. Each component of ADCs, including antibodies, linkers, and cytotoxins, require independent optimization, and any minor modifications in these key components can lead to substantial changes in clinical characteristics. Therefore, in designing future BsADCs, the optimization of the antibody, linker-cytotoxin complex, and conjugation strategy should be viewed as interconnected networks, requiring a holistic approach.
- Selection of bispecific antibodies
The primary consideration in constructing BsADCs is to wisely select the appropriate target combination. Target selection is a fundamental prerequisite for the successful development of ADC, having a key impact on the final therapeutic window and systemic toxicity. Given the widespread off-target toxicity and clinical resistance challenges faced by traditional ADC, the following criteria can help guide target selection:
(1) Traditional target selection is based on good internalization characteristics, relatively low expression in normal tissue, and high expression in tumors.
(2) Given the heterogeneity of solid tumors, the levels of target expression in various tumor subtypes and locations must be determined, which promotes the optimal implementation of tailored drug delivery instead of relying on a single “panacea”.
(3) Due to the unique dual-targeting characteristics of BsADCs, it is critical to fully consider the deeper effects of the combination of antigens, including internalization, recirculation, turnover rates, lysosomal degradation, and inherent mechanisms, among other factors. The integration of these factors is essential for the effective design of BsADCs.
In addition, one of the main classification criteria for BsADCs is whether they contain an Fc region. The design of BsADCs without Fc faces challenges such as low stability, aggregation problems, and a lack of coupling sites. On the other hand, BsADCs with Fc bring additional advantages, such as ADCC, CDC, immune phagocytosis, and cytokine release, all of which contribute to tumor killing. In summary, the strategies for Fc region construction include:
- Fc engineering modifications, such as amino acid mutations and glycosylation modifications in the Fc region, which can help alleviate off-target toxicity caused by FcγR binding.
- Retaining ADCC and CDC: the dual target binding mode promotes the formation of hexamers, which can enhance ADCC and CDC activity, and improve tumor killing effects.
- Retaining FcRn binding or applying antibody engineering can help to improve the half-life and safety.
- Selection of linkers
In BsADCs, the linker plays a pivotal role in the release of payloads and the stability of the drug. Ideally, the linker should display stability in plasma while promoting effective release in tumors. Currently, linkers in ADCs can be categorized into cleavable and non-cleavable linkers.
Non-cleavable linkers exhibit a high degree of stability in plasma and can release payloads through lysosomal degradation only. This type of linker leads to lower off-target toxicity, increased plasma half-life, and enhanced safety. However, potential resistance could stem from obstacles in internalization and lysosomal transport. BsADCs based on non-cleavable linkers should focus on further internalization optimization, as well as subsequent endosome transport and lysosomal degradation.
Compared to non-cleavable linkers, ADCs based on cleavable linkers have a broader application. The main challenge for cleavable linkers is off-target toxicity due to non-specific release. To design lysosome-independent BsADCs, certain cleavable linkers (such as Val-Cit linkers) can effectively promote payload release from early and late endosomes. In addition, the concept of non-internalizing ADCs has been proposed recently, where chemical or enzyme-mediated cleavage is triggered extracellularly, allowing targeting of antigens in the TME and vascular system, and activating bystander killing effects.
Our Hot Linker Products
| Catalog | Product name | CAS NO | Inquiry |
| ADC-L-003 | Fmoc-Val-Cit-PAB-OH | 159858-22-7 | Inquiry |
| ADC-L-004 | Fmoc-Val-Cit-PAB-PNP | 863971-53-3 | Inquiry |
| ADC-L-005 | Mc-Val-Cit-PABC-PNP | 159857-81-5 | Inquiry |
| ADC-L-007 | Val-Cit-PAB | 159857-79-1 | Inquiry |
| ADC-L-008 | MC-Val-Cit-PAB-PNP | 159857-81-5 | Inquiry |
| ADC-L-016 | SMCC | 64987-85-5 | Inquiry |
- Selection of Payload
The cytotoxic payload significantly determines the overall antitumor effect and potential adverse reactions. Given the low penetration of ADCs, an ideal ADC payload needs to demonstrate high potency at the nanomolar to picomolar level. In addition, these effective payloads should have sufficient plasma stability, low immunogenicity, and appropriate water solubility. Finally, the effective load should have available groups for coupling with antibodies.
The bystander killing effect of the effective payload in BsADCs is a crucial aspect worth discussing, which refers to the ability of an effective payload to kill adjacent non-target cells after release. For the PK/PD characteristics of ADCs, it is a double-edged sword. Although the bystander killing effect can enhance the overall efficacy of ADCs in a heterogeneous tumor environment, it also poses the risk of off-target killing in normal tissue around the tumor. This effect depends on cleavable linkers and hydrophobic effective payloads. If one of the target antigens has a certain level of expression in normal tissues (such as c-Met), BsADCs should avoid administering payloads with bystander effects. In summary, the bystander killing effect of BsADCs is expected to overcome tumor heterogeneity, tumor barriers, and poor internalization. However, potential safety issues related to off-target effects need to be carefully considered.
Additionally, the development of novel drug payloads is crucial for the development of BsADCs. Emerging payloads, such as PROTACs, ferroptosis inducers, and oligonucleotides, can aid in expanding drug selection in the BsADC field. The development of new drugs can significantly enrich the selection of selective drug types in the BsADC field.
BsADC targeting HER2
ZW49
ZW49 is based on Zanidatamab, using the interchain disulfide bond cysteine and protease cleavable linker to couple N-acylsulfonamide auristatin, making it well-tolerated. The bispecific antibody properties of ZW49 contribute to better internalization, and its Fc region imparts ADCC, ADCP, and CDC effects. This design addresses several unmet clinical needs of HER2-expressing patients. Preclinical data showed that ZW49 exhibited a strong tumoricidal effect and good tolerance without compromising HER2 affinity. Currently, ZW49 is undergoing Phase I clinical trials. As of September 2022, disclosed clinical trial data show that the objective response rate (ORR) in patients with HER2-expressing advanced solid tumors is 31%, and its ocular toxicity feature (corneal keratitis as 42%) is notable.
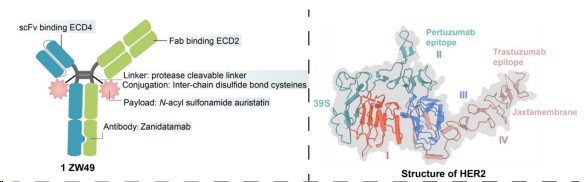
Fig.2. Structure of ZW49 (Gu Y,2024)
MEDI4276
MEDI4276 is a tetravalent HER2-targeted ADC that fuses an scFv of trastuzumab with the N-terminal of another anti-HER2 IgG1 antibody 39S. MEDI4276 demonstrated significant activity in mouse xenograft models of refractory HER2+ cancers but did not display a good therapeutic efficacy-safety balance in clinical trials. In patients with breast cancer, the overall ORR was low (9.4%), and the maximum tolerated dose (MTD) was established at 0.75 mg/kg every three weeks. The lower MTD of MEDI4276, compared to ZW49, may be influenced by its valency, effective load, and antibody configuration, indicating a need for further optimization.
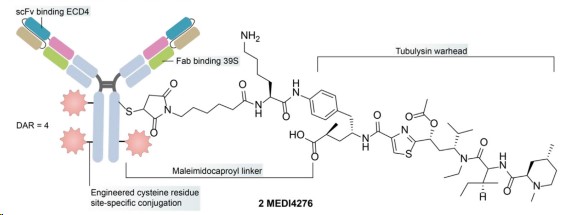
Fig.3 Structure of MEDI4276 (Gu Y,2024)
BsADC targeting EGFR
EGFR is a member of the ERBB receptor tyrosine kinase family, playing a crucial role in regulating the basic functions of epithelial malignancies. However, due to treatment pressure-induced acquired genomic changes, targeting EGFR monoclonal antibodies and TKIs often leads to the emergence of clinical resistance. BsADCs have the potential to address anti-EGFR resistance mechanisms, including sensitizing mutations and the activation of bypass pathways.
BsADC targeting EGFR double epitopes
In order to alleviate the emergence of drug resistance, a bispecific antibody against two different epitopes of EGFR has been developed. This bispecific antibody is designed by fusing nano-antibodies specific to non-overlapping epitopes (9G8 and 7D12) on EGFR.
7D12 disrupts the EGFR signaling cascade, while 9G8 stabilizes the binding conformation of EGFR-ECD, spatially preventing dimerization. Furthermore, 7D12 and 9G8 demonstrate potency against different EGFR mutant cell lines, inducing more effective CDC effects in NIH-3T3 cells that express wild-type EGFR or show resistance mutation to cetuximab.
BsADCs targeting EGFR and other antigens
So far, several BsAbs targeting c-MET and EGFR have been reported, demonstrating their synergistic effects in inhibiting tumor proliferation and metastasis. In the design of BsADCs, careful selection of the right epitope combination is crucial to avoid complete or partial agitation of c-MET. AZD9592 is an EGFR/c-Met ADC developed by AstraZeneca, coupled with a novel topoisomerase I payload via a cleavable linker, mainly addressing resistance to osimertinib. Compared to EGFR, AZD9592 has a higher affinity for c-MET, aiming to reduce normal tissue toxicity driven by EGFR. In PDX and resistance models, it demonstrated good antitumor activity either alone or in combination with osimertinib.
1231 is a MUC1/EGFR bispecific ADC developed in collaboration by Sutro and Merck subsidiary EMD Serono. It uses non-natural amino acid site-specific coupling technology, coupling a hemiasterlin derivative (microtubule inhibitor) via a cleavable VC linker, with a DAR of 4. Preclinical studies show strong antitumor activity in patient-derived xenograft models of ESCC and NSCLC.
BsADC targeting MET
The hepatocyte growth factor (HGF)-mesenchymal-epithelial transition factor (MET) pathway plays a crucial role in the development of various types and stages of cancer, from initiation to metastasis. Upregulation and amplification of MET are regarded as major escape mechanisms during anti-EGFR therapy. C-MET can cross-react with EGFR, leading to resistance to EGFR targeted therapy, thus making the inhibition of C-MET a feasible strategy to overcome EGFR resistance.
Compared to traditional MET-targeted ADCs, the design of bispecific MET×MET-BsADC offers an innovative solution to overcome existing challenges. The bispecific antibody to MET has the ability to form a 2:2 antigen-antibody complex, promoting effective MET internalization and lysosomal transport. By coupling the Maytansinoid effective payload M114 to the lysine on the surface of the MET-target BsAb via a protease-cleavable linker, REGN5093-M114, a BsADC with a drug-to-antibody ratio (DAR) of 3.12 is generated.
Preclinical data show that REGN5093-M114 significantly inhibits the proliferation of MET-overexpressing NSCLC cells. A Phase I, dose-escalation, and expansion study to assess the safety and efficacy of REGN5093-M114 in adult patients with MET-overexpressing advanced cancers has now been initiated (NCT04982224).
New ADC
BsADCs significantly expand the range of potential targets and scaffolds beyond traditional paradigms. In fact, BsADCs provide an efficient means to transform non-internalizing antigens into internalizing ones. For example, generating a BsAb by utilizing EphA2, characterized by rapid internalization, and activated leukocyte adhesion molecule (ALCAM), a non-internalizing or slowly internalizing antigen. Interestingly, when the cellular surface ratio of EphA2 to ALCAM exceeds the threshold value of 0.2, the bispecific antibody demonstrates effective internalization. Conversely, when the ratio falls below this threshold, internalization is hindered.
The MC-VC-pab-MMAF payload complex is site-specifically linked via cysteine residues. In the context of bispecific binding of BsADCs, the internalization effect can be significantly influenced by simultaneously targeting adjacent antigens, depending on their expression ratio. This ability expands the range of antigen selection and can be used to design BsADCs with poor internalization. This innovative approach provides a subtle strategy for enhancing the internalization potential of certain antigens, contributing to the multifunctionality and effectiveness of BsADCs targeting a wider range of tumor antigens.
The introduction of various new technologies has completely changed the generation of BsADCs, presenting obvious advantages. Another significant advancement is the PEG-linked BsADC (P-BsADC), which not only ensures uniform coupling production but also demonstrates high endocytosis efficiency, tissue penetration, and reduced toxicity due to its small molecular weight and the absence of Fc fragments.
In addition, ligand-induced transient engagement (LITE) of multiple antibody structural domains is a cutting-edge technology that combines the half-life extension advantages of biopharmaceuticals with precise temporal control of small molecule relevant activities. These innovative strategies have great prospects for achieving therapeutic effects while minimizing off-target effects, representing an important step in the development of BsADC technology.
In conclusion, the emergence of unique bispecific targeting models brings new innovation to the ADC field, marking the birth of the new generation of ADCs. Although in the early stages of development, BsADCs offer a new approach with considerable potential. BsADCs are of significant importance for overcoming the existing clinical challenges faced by traditional ADCs and for developing more precise, targeted drugs.
Reference:
Gu Y, Wang Z, Wang Y. Bispecific antibody drug conjugates: Making 1+ 1> 2[J]. Acta Pharmaceutica Sinica B, 2024.