
A Simple Concept of Antibody-drug Conjugates (ADCs)
Developing new anticancer chemotherapeutic drugs showing superior antitumor efficacy with reduced toxicity is continuing to be a challenging endeavor. Antibody-drug conjugates (ADCs) are an emerging class of targeted anticancer drug delivery agent that confer selective and sustained cytotoxic drug delivery to tumors. More than a century ago, in 1906, Paul Ehrlich came up with the original concept of linking a cytotoxic payload to a protein with affinity and selectivity to a tumor, like an antibody (Ab) to achieve selective delivery of chemotherapeutic agents in a tumoral environment. Thus, the idea of antibody–drug conjugates (ADC) was born (Figure. 1).
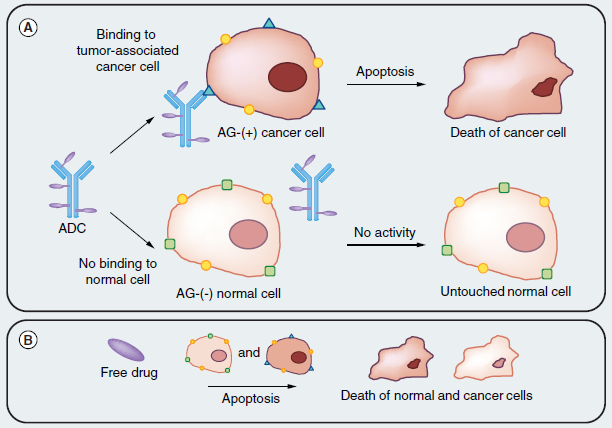
Figure. 1 Basic early antibody-drug conjugate concept illustration. (A) An antibody specific to a tumor-associated antigen recognizes the cell surface of a tumor cell, leaving normal tissue untouched. Then, drug release and action through various mechanisms leading to cell death. (B) In comparison, the free drug is toxic against both normal and cancer tissues, therefore, showing no selectivity between tumors and healthy tissue.
Many monoclonal Ab (mAb), such as avastin, rituximab, and cetuximab, are well known as standard treatments for solid tumors and hematological cancers. By contrast, pristine chemodrugs, such as vinblastine, doxorubicin, and paclitaxel, have limited use for cancer treatment because of their nonspecific toxicity, thus narrowing the therapeutic window and increasing drug resistance. Discovery of ADCs bridged the gap between the Ab and cytotoxic drug, creating highly specific anticancer agents with an improved therapeutic window. This simple concept was thought to be a particularly attractive solution to the challenge of finding a way to increase the therapeutic window of the cytotoxic agent. Furthermore, conjugation of a small molecular weight cytotoxic agent to a large hydrophilic antibody is expected to restrict penetration of the cytotoxic compound across cellular membranes of antigen-negative normal cells, providing an additional mechanism by which the therapeutic index of the small molecule cytotoxin is widened, beyond that of targeted delivery (Figure. 2).
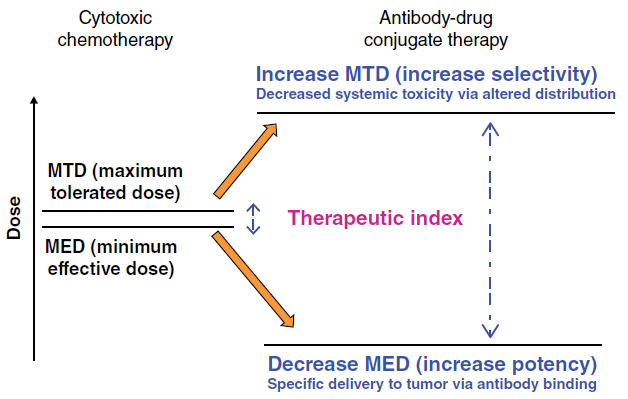
Figure. 2 Increasing the therapeutic index of cytotoxic drugs by conjugation to antibodies.
Advances in ADC Development
First-generation ADCs
Since the breakthrough discovery of the first FDA-approved ADC, gemtuzumab ozogamicin (trade name, Mylotrag), anticancer therapy research has diversified, resulting in the development of several novels, safer ADCs. Advances in the development of ADCs towards targeted anticancer drug delivery is described by Creative Biolabs here. After the first generation of ADC had caused acute adverse effects and morbidity in patients, several parameters were pointed out that cannot be bypassed while developing an ADC:
What We Learned From First-generation ADCs
- Insufficient potency of ADC in patients: due to low concentrations of antibody both in serum and moreover in tumor environment. The low concentration of mAb in plasma cannot allow to achieve the appropriate therapeutic range, and only <1% of the injected dose is able to reach the tumor localization.
- Nature of the antibody: nonhuman antibodies were used in first ADC, leading for example to an immune response through the generation of human anti-murine antibodies (HAMA), preventing repeated exposure of the ADC to patients, thus limiting its efficacy.
- Linker stability: first linkers were designed to be cleaved under acidic conditions, nevertheless this kind of linker proved to be quite unstable even in plasma, leading to unexpected toxicity due to the early uncontrolled release of the toxic payload.
- Limited expression of the antigen: before delivering and releasing the payload inside the cancer cell, the ADC should first bind to its specific antigen at the tumor cell surface. Because of the limited expression of this antigen at the surface, which should be of an average of 10,000 receptors per cancer cell, the ADC can only deliver a limited number of payloads that could be insufficient to reach the necessary concentration to achieve cell death.
- Limited internalization: not all the targeted antigen can allow an optimal clathrin dependent internalization of the antigen-mAb complex.
- Optimized average DAR = 4: this value should be the best one to achieve in order to build an optimized ADC, having in mind the compromise between a sufficient number of carried drugs and a limited modification of the native structure of the mAb.
Second-generation ADCs
The limitations and failures of first-generation ADCs were eliminated in second-generation ADCs. The premature release of drugs because of the unstable hydrazone linker in Mylotrag® has been avoided in second-generation FDA approved ADCs, by using different linkers, such as the valine-citrulline (cathepsin cleavable) linker in Adcetris® and a thioester (noncleavable) linker in Kadcyla ®. The cytotoxic payloads used in second-generation ADCs are also more potent than in first-generation ADCs. For example, tubulin-targeting agents, such as MMAE used in Adcetris® is approximately 100–1000-fold stronger than DNA-intercalating doxorubicin of BR96 Dox. The IC50 of MMAE is approximately 1 nM in different human cancer cell lines, whereas doxorubicin IC50 is in the 1–6 uM range. Despite the improvement in cytotoxic payloads and the introduction of stable linkers, second-generation ADCs have significant limitations in terms of their heterogeneous DAR, resulting from stochastic coupling strategies between the Ab and drug. Typically, chemical conjugation between the drug and antibody occurs via the lysine or cysteine residue of the mAb, which generates DAR (range 0–8) with an average value of 3–4. Therefore, heterogeneous ADCs can contain a mixture of unconjugated, partially conjugated, and overconjugated antibodies and, therefore, there will be competition between unconjugated antibodies and drug-conjugated species for antigen binding that can diminish the activity of the ADC. By contrast, overconjugation of the drug to the Ab can result in antibody aggregation, a decrease in stability, and incremental increases in nonspecific toxicity, and a reduction in the half-life of ADCs in the circulation. Overall, heterogeneous ADCs have a limited therapeutic index and tumor penetration abilities, which are associated with the induction of drug resistant in the tumor microenvironment.
What We Learned From Second-generation ADCs
- Mode of action and potency of the payload: only DNA alkylating agents and tubulin polymerization inhibitors with subnanomolar activities proved to be useful for targeted delivery through ADC technology, due to limited antigen expression and limited delivered amount of ADC available in the tumor. Such drug cannot be used in monotherapy due to high cytotoxicity and narrow therapeutic window.
- Nature of the linker and delivery mechanism: cleavable linkers finally have a broader efficacy than noncleavable ones. Indeed, although ADC incorporating either cleavable or noncleavable linker requires internalization to release the free drug, ADCs with cleavable linkers may also be active even when they are poorly internalized. The explanation can be found in the overexpression of some intracellular enzymes, like cathepsin B, that can leave lysosomes then cytosol to reach the extracellular media to promote metastasis or cell invasion, leading to cleavage of the linker in extracellular matrix, the free drug subsequently permeates the cell to reach its target [16].
- Conjugation site on the mAb: it can also affect potency, stability and PK properties of the ADC.
- Parameters to choose a good AG target: homogeneity/heterogeneity of tumor expression, high level of expression inside a tumor and selective expression for tumor tissues should be all well assessed for a given AG to be considered as a suitable target for ADC payload delivery.
Third-generation ADCs
The aforementioned concerns regarding the heterogeneous DARs of second-generation ADCs have been addressed in third-generation ADCs. Site-specific conjugation has been introduced to produce homogenous ADCs with well-characterized DARs and desired cytotoxicities [62]. The site-specific conjugation of the drug to Ab provides a single isomer ADC with a uniform DAR value. Such ADCs can be made using bioengineered antibodies containing site-specific amino acids, such as cysteine, glycan, or peptide tags [63]. For example, precise site-specific conjugation of MMAE to human IgG was developed by replacing the Ala114 amino acid of the CH1 domain of the IgG with cysteine to create a selectively engineered antibody, called THIOMAB. Alternative approaches to site-specific drug conjugation include: (i) a thio-bridge approach that links drugs to the interchain disulfide bond of Abs (four per mAb) [14]; (ii) introduction of unnatural amino acids, such as p-acetylphenylalanine, or noncanonical amino acids. Based on above techniques, Creative Biolabs has developed a novel ADC conjugation platform-PtLnX™. PtLnX is a technical platform developed by Creative Biolabs and one of our collaborators that uses bis(ethylenediamine) platinum chloride [Pt(en)Cl2] as a linker core for ADC development.
Wisely chosen targeted antigen, novel linker technology and original mode of drug action continue to be investigated to fully optimized ADC-based targeted therapy in order to improve the therapeutic index of an overall ADC. Extensive research is ongoing to improve all the components of ADCs that can enhance their targetability and therapeutic efficacy against tumors. A better understanding of ADC-targeting strategies can speed up the FDA approval rate of ADCs and drastically increase the number of clinical trials, especially in solid tumors.