T cell receptors (TCRs) provide a recognition signal for T cells complemented by a co-stimulatory signal that can provide an on/off signal to regulate the activation of T cells. Since Medawar and colleagues carried out their seminal work, it has long been recognized that adoptively transferred T cells have the potential to target and destroy cancer cells which brings us the concept of T cell therapy. The earliest clinical trials of engineered T cells in cancer relied on the expression of cloned TCRs with targeted affinity for tumor antigens. More recently, artificial receptors, such as chimeric antigen receptors (CARs), have been used to enhance engineered T cell specificity. CARs are formed from a combination of antibody-derived or ligand derived domains as antigen recognition part and TCR domains as signal transduction part. Adoptive transfer of T cells expressing engineered receptors has shown enormous promise in humans. With the proof of concept established, engineered T cells have matured as a potential therapeutic option to treat malignancies. Building on this foundation, the field is broadening indications for current therapies, exploring new targets and using new techniques to create even safer and more effective therapies.
Here we recommend a review, which describe some of the most recent and promising advances in engineered T cell therapy, with a particular emphasis on what the next generation of T cell therapy is likely to entail.
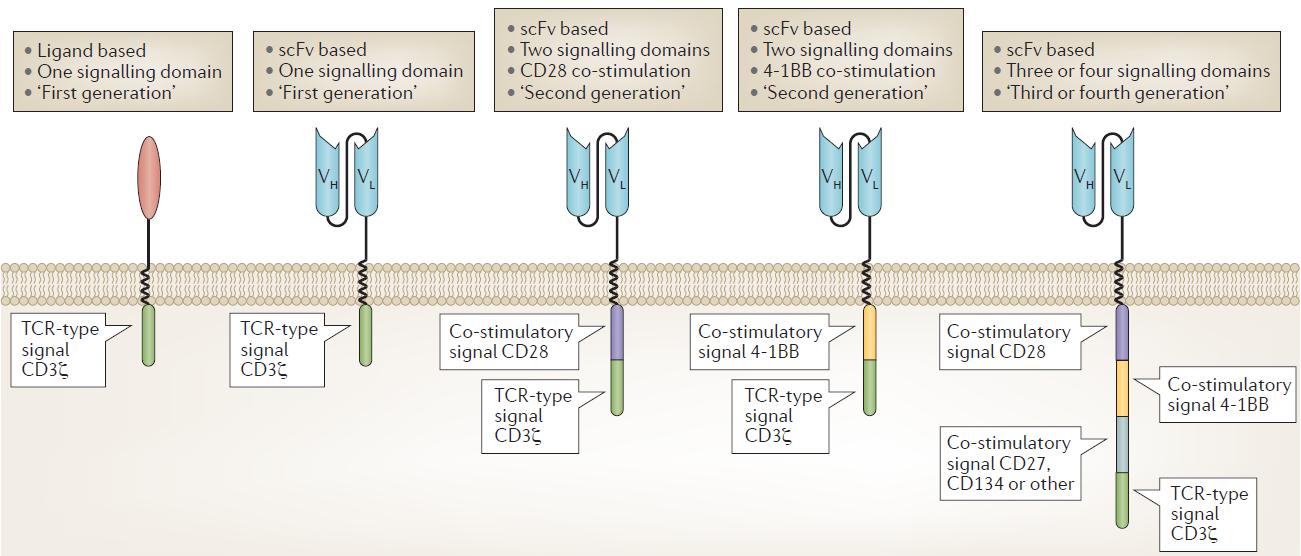
Figure 1. Chimeric antigen receptor design and evolution (Fesnak, 2016)
The first clinical trials tested CARs that had a binding domain composed of native CD4 that bound to the envelope glycoprotein gp120 expressed by HIV-infected cells, with a single T cell receptor (TCR) signaling domain composed of the CD3ζ chain. CARs with an extracellular domain composed of antibody single-chain variable fragments (scFvs) were first reported by
Kuwana and later by Eshhar and colleagues. Second-generation CARs incorporating CD28 as a co-stimulatory domain were first developed by Roberts and reported by Finney, and those incorporating 4‑1BB as a co-stimulatory domain were developed and reported by Finney, Imai and then others. CARs incorporating three or four signaling domains, so called ‘third- and fourth-generation’ CARs, have also been developed and are beginning to be tested in clinical trials. CAR T cell design has evolved over the years to enhance efficacy and safety in particular immunological settings.
Clinical feedback has enabled re‑evaluation of some basic tenets of CAR T cell targeting. Whereas previous approaches emphasized efficacy, minimization of off-tumor effects is now the primary driver of target selection when potent CAR T cells are used. CAR T cells are able to respond to minimal target expression, making target specificity particularly important. Off-tumor effects can be lethal and currently limit clinical applications, particularly with regard to solid tumor therapy.
Finally, new findings force us to rethink what it means for a T cell therapy to be ‘specific’ for a target. Two-step approaches are being used, wherein T cells are engineered to express a receptor with affinity for a non-specific molecule and this molecule is then fused to a targetable agent with high affinity for the tumor. For example, Kim et al. created a fluorescein isothiocyanate (FITC)-specific CAR T cell, which bound to a fusion FITC–folate molecule. Folate receptor (FR)+ cells are then ‘painted’ with the FITC–folate molecule, enabling FITC-CAR T cells to target FR+ cells.
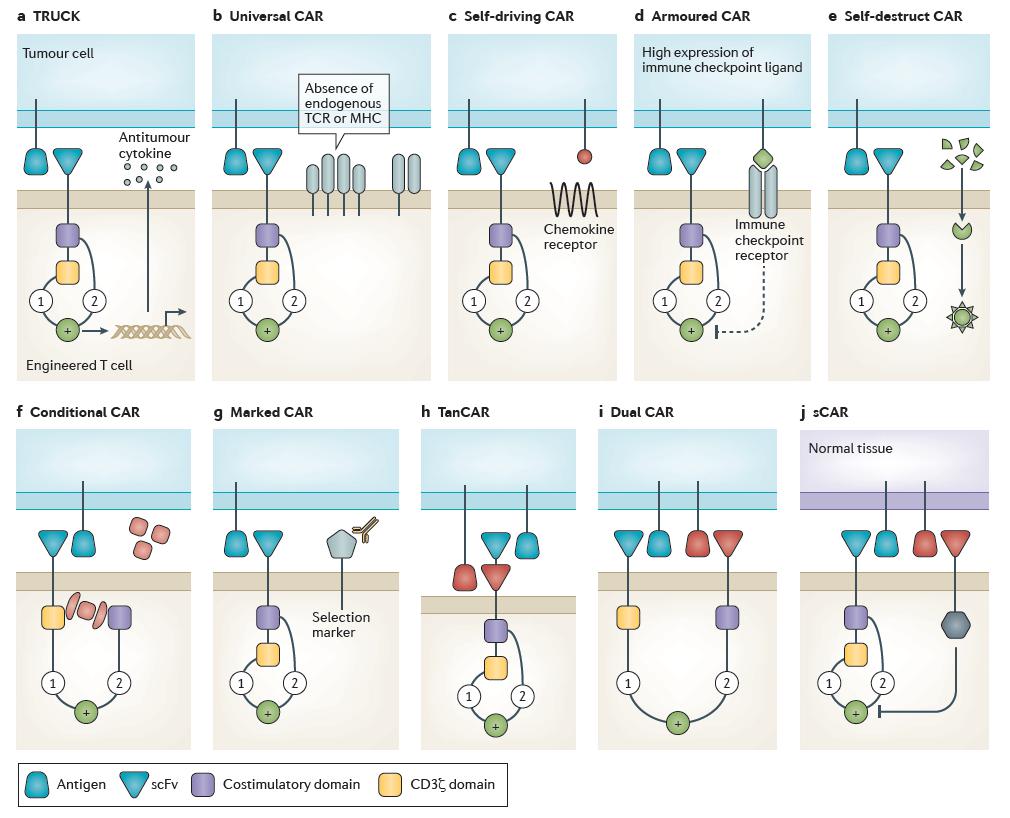
Figure 2. New chimeric antigen receptor models and concepts. (Fesnak, 2016)
TRUCK CAR
Scientists have generated T cells specific for tumor antigen that, upon binding, produce cytokines that are intended to recruit endogenous immune cells and mediate tumor clearance. This kind of CAR T cell design is called T cells redirected for universal cytokine killing (TRUCKs). TRUCKs co‑express a chimeric antigen receptor (CAR) and an antitumor cytokine. Cytokine expression may be constitutive or induced by T cell activation (for example, interleukin‑12 (IL‑12)). Targeted by CAR specificity, localized production of pro-inflammatory cytokines recruits endogenous immune cells to tumor sites and may potentiate an antitumor response.
Universal CAR
Gene editing is one of the most exciting recent developments in the modernization of redirected T cell manufacturing. Current gene modification techniques used to produce engineered T cells must balance efficiency, safety and cost. Efficient gene editing of primary human T cells has been demonstrated using ZFNs, TALENs and CRISPR–Cas9. The manufacture of gene-edited human CAR T cells has been shown to be feasible with the production of TALEN or CRISPR–Cas9 gene-edited CAR T cells. Allogeneic CAR T cells are engineered to no longer express endogenous T cell receptor (TCR) and/or major histocompatibility complex (MHC) molecules, thereby preventing graft-versus-host disease (GVHD) or rejection, respectively. Although promising, this exciting step forward will require further investigation in more patients to demonstrate the role of allogeneic CAR T cells in tumor control.
Self-driving CAR
Engineered T cell localization at target sites is crucial for clinical efficacy, particularly when targeting solid tumors. Although T cells can migrate to nearly all body compartments, including immune privileged sites accumulation of engineered T cells may be enhanced by local administration. Chemokine receptor–ligand interactions have an important role in mediating endogenous immune cell trafficking. In fact, the efficacy of conventional chemotherapeutics is linked to the upregulation of chemokine ligands on tumour cells that is mediated by these drugs CAR T cells may be engineered to express chemokine receptors to enhance trafficking into tumor tissue and homing to tumor cells. Self-driving CARs co‑express a CAR and a chemokine receptor, which binds to a tumor ligand (for example, C‑C motif chemokine receptor 2 (CCR2)–C‑C motif chemokine ligand 2 (CCL2)), thereby enhancing tumor homing.
Armored CAR
Malignancy may be refractory to engineered T cell therapy owing to immune escape or immunosuppression in the tumor microenvironment. Various methods can be used to engineer T cells to be intrinsically resistant to tumor immunosuppression. CAR T cells engineered to be resistant to immunosuppression (armored CARs) may be genetically modified to no longer express various immune checkpoint molecules (for example, cytotoxic T lymphocyte-associated antigen 4 (CTLA4) or programmed cell death protein 1 (PD1)), with an immune checkpoint switch receptor, or may be administered with a monoclonal antibody that blocks immune checkpoint signaling. Future T cell therapies are likely to incorporate multiple forms of immune checkpoint blockade to further enhance efficacy.
Self-destruct CAR
unexpected and fatal cross-reactivity seen with engineered TCR T cells demonstrates the current limitations of in vitro screening for cross-reactivity. Molecular ‘switches’ enable greater control over the performance of engineered T cells in vivo and may improve safety. Cells may be engineered to express pro-death signals that can be induced with an exogenous element. A self-destruct CAR may be designed using RNA delivered by electroporation to encode the CAR.
Conditional CAR
T cells may be engineered to conditionally activate only in the presence of an exogenous molecule, withdrawal of which terminates signaling. A conditional CAR T cell is by default unresponsive, or switched ‘off’, until the addition of a small molecule to complete the circuit, enabling full transduction of both signal 1 and signal 2, thereby activating the CAR T cell.
Marked CAR
Engineered T cells may be marked with unique cell surface molecules to which approved therapeutic monoclonal antibodies bind. If this epitope is also expressed on tumor cells, treatment with these monoclonal antibodies could eliminate CAR T cell-mediated adverse effects while simultaneously treating the tumor. Marked CAR T cells express a CAR plus a tumor epitope to which an existing monoclonal antibody agent binds. In the setting of intolerable adverse effects, administration of the monoclonal antibody clears the CAR T cells and alleviates symptoms with no additional off-tumor effects.
TanCAR
Deletion of CAR T cells may limit adverse effects, but will also terminate the antitumor clinical effect. Off-tumor toxicity can also be prevented by designing CAR T cells with enhanced specificity. To achieve this, CAR T cells have been designed to activate only in response to a particular combination of targets. For example, bispecific CARs have been generated such that the extracellular portion of the CAR contains two linked scFvs with different specificities. T cells expressing these tandem CARs (TanCAR) are activated only in the presence of both targets; a target cell positive for a single antigen is insufficient to trigger T cell activation and cell killing.
Dual CAR
A dual CAR T cell expresses two separate CARs with different ligand binding targets; one CAR includes only the CD3ζ domain and the other CAR includes only the co-stimulatory domain(s). In this approach, a single T cell expressing a CD3ζ‑only CAR against the first target and a co-stimulatory domain-only CAR against a second target will become fully activated only in the presence of both targets.
sCAR
Lastly, extracellular scFv fused to inhibitory signaling domain can specifically inhibit CAR T cell activation. These inhibitory signals enable protection of cells with particular immunophenotype from CAR T cell killing. A safety CAR (sCAR) consists of an extracellular scFv fused to an intracellular inhibitory domain (for example, CTLA4 or PD1). sCAR T cells co-expressing a standard CAR become activated only when encountering target cells that possess the standard CAR target but lack the sCAR target.
All of these special design of CAR T cell may be applied to the next generation of engineered TCRs to improve targeting and control. The future development tendency of CAR design may be more and more focus on the safety and controllable issue of this kind of new therapy. Although there are some promising results in CAR T therapy clinical trials, much more effort should be taken until CAR T technology becomes a mature, stable and more safe therapy strategy.
Reference
Fesnak, Andrew D., Carl H. June, and Bruce L. Levine. “Engineered T cells: the promise and challenges of cancer immunotherapy.” Nature reviews cancer 16.9 (2016): 566-581.