Duchenne muscular dystrophy (DMD) is a monogenic disease with a relatively high prevalence, caused by mutations in the gene encoding the anti-myotonic protein (Dystrophin) on the X chromosome, resulting in the failure to produce enough of the anti-myotonic protein and the progressive replacement of muscle tissue with fat and fibrotic tissue, which affects approximately 1 in 5,000 newborn boys.
In recent years, several antisense oligonucleotide (ASO) drugs have been approved for the treatment of DMD by exon skipping, but these drugs only cover a small number of patients with DMD at specific mutated loci.
Gene replacement is a better alternative to exon skipping, However, the Dystrophin gene is so large, with up to 79 exons and up to 14kb long transcripts, which is well beyond the loading limit of viral delivery vectors, making direct delivery of the correctly encoded Dystrophin gene unfeasible as a therapeutic approach. To address this challenge, a miniature Dystrophin gene has been developed that is still functional and small enough to be delivered using an adeno-associated virus (AAV) vector.
Recently, researchers from Texas A&M University, the University of Florida, and Solid Biosciences published a paper in the journal Science Translational Medicine entitled “Assessment of systemic AAV-microdystrophin gene therapy in The GRMD model of Duchenne muscular dystrophy”.
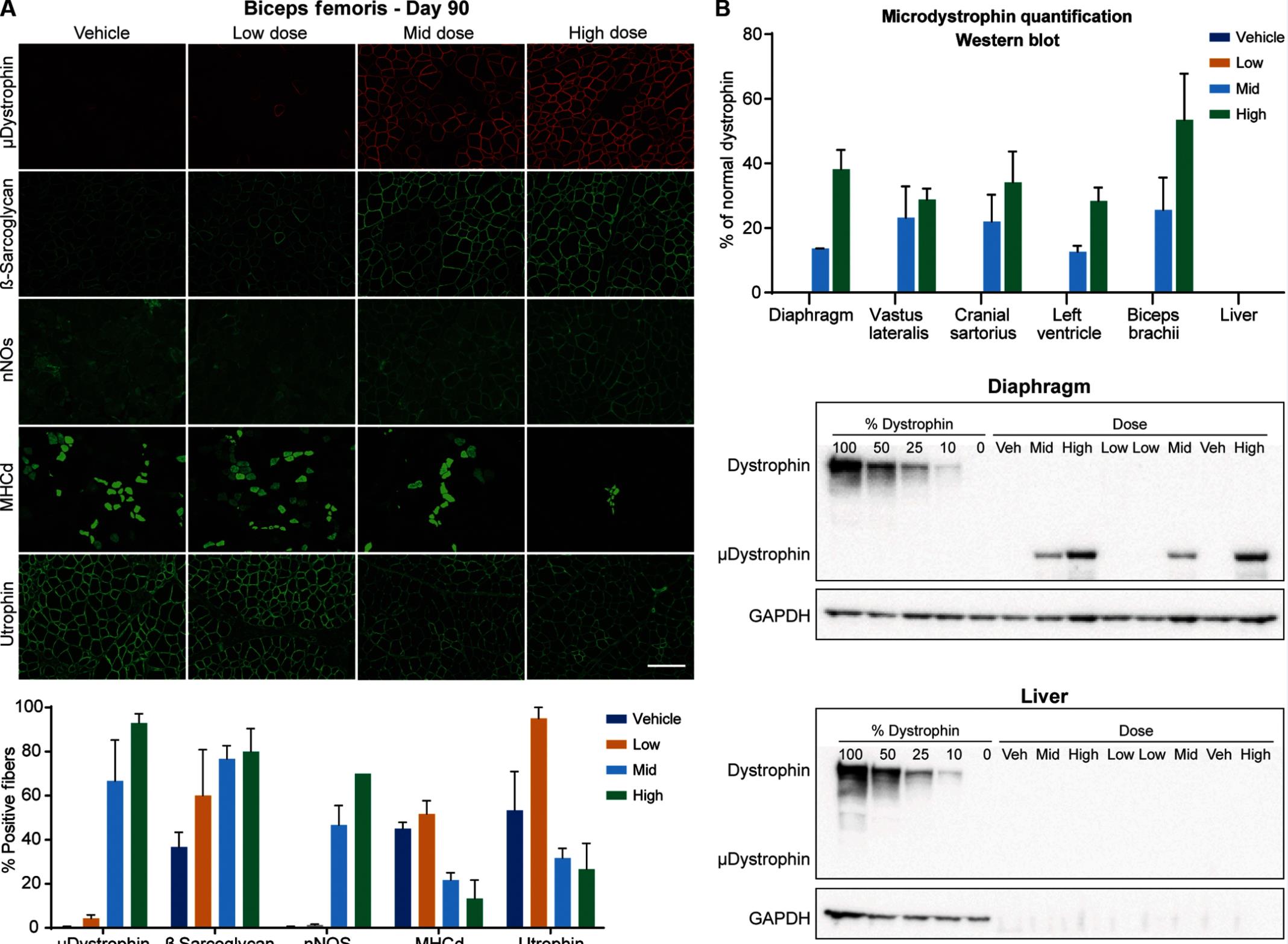
The study showed that AAV-delivered microdystrophin gene therapy improved symptoms in the golden retriever muscular dystrophy (GRMD) model without serious adverse events. This suggests that the administration of Dystrophin protein via the AAV system may be a safe gene therapy that provides therapeutic benefit to patients with DMD.
Studies of DMD are usually conducted on mouse models of DMD, but the results of studies on large animals such as the GRMD model are more readily translatable to humans.
In this study, the team used AAV type 9 (AAV9) to deliver the microdystrophin gene (μDys5) for preclinical studies in the GRMD model. Twelve GRMD dogs were randomly divided into four groups—control group, group with 1 x 10E13vg/kg dose, group with 1 x 10E14vg/kg dose, and group with 2 x 10E13vg/kg dose—with three dogs in each group.
The miniature Dystrophin gene (μDys5) delivered by intravenous injection of AAV9 was administered to these dogs at 3 months of age and followed up for 90 days after administration. All dogs received prednisone (at a dose of 1 mg/kg, a DMD hormone therapy) for a total of 5 weeks from day 7 to day 28.
It is worth noting that previous gene therapy studies have typically used cytomegalovirus (CMV) promoters to initiate expression of target genes, but CMV promoters may initiate higher levels of expression in non-muscle tissues and generate associated immune responses. To reduce the immune response to the microdystrophin protein, this study provided proof of concept showing the use of a muscle-specific promoter that initiates expression of the microdystrophin protein in muscle tissue only, and showed that GRMD dogs exhibited sustained protein expression for 8 weeks in the absence of immunosuppression.
The results showed a dose-based increase in genomic copy number in the tissues of treated dogs, expression of the μDys5 protein in muscle tissue and heart, improved limb and respiratory muscle function, and reduced histopathological lesions in the dogs. As expected, the phenotype and histopathological lesions did not fully return to normal due to the expression of the microdystrophin protein. In addition, the different dose treatment groups were well tolerated with no adverse events.
These data suggest that systemic administration of AAV-delivered microdystrophin proteins is safe and may provide therapeutic benefits to patients with DMD.
Reference
1. Birch, Sharla M., et al. “Assessment of systemic AAV-microdystrophin gene therapy in the GRMD model of Duchenne muscular dystrophy.” Science Translational Medicine 15.677 (2023): eabo1815.