The success of CAR T cells in treating hematologic malignancies has provided impetus for the development of CAR T therapies in solid tumors. The biggest obstacle to the development of CAR T therapies in solid tumors is that most candidate antigens are co-expressed on non-malignant tissues, which can lead to on-target off-tumor toxicity (OTOT) and greatly elevate the risk of adverse effects. Preclinical and clinical studies using CAR T cells targeting antigens shared by malignant and non-malignant tissues have reported varying degrees of OTOT responses, making overcoming OTOT effects the greatest challenge in applying CAR T cell therapies to solid tumors.
Recently, Professor Mohamed Abou-el-Enein’s team from the Keck School of Medicine of the University of Southern California (USC) published an article entitled “Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumors” in Nature Reviews Clinical Oncology. In this review, they provide insight into the impact of OTOT on the development of CAR-T cell therapy for solid tumors, summarize evidence from preclinical and clinical studies of OTOT, and discuss recent advances in CAR-T research that may help overcome the effects of OTOT in the clinic.
Risks And Mechanisms of OTOT
OTOT arises from the recognition and lysis of non-malignant tissues expressing target antigens by CAR-T cells, a response that can lead to serious adverse effects. Once the target antigen is recognized, CAR-T cell activation leads to the formation of an immune synapse between CAR-T cells and the target cell, triggering an effector effect. The release of perforin and granzyme is the main mechanism of CAR-T cell-mediated cytotoxicity. However, other mechanisms may also lead to tissue destruction, such as upregulation of T cell surface molecules to induce apoptosis in target cells (e.g., FAS ligands) or secretion of cytokines, including IFNγ and/or TNF.
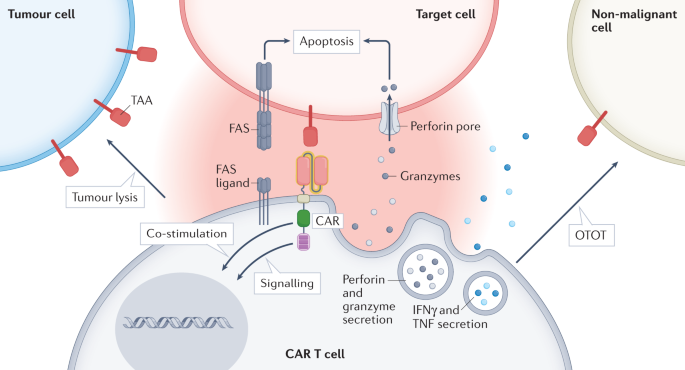
In order to generate CAR T cells that are both safe and effective for patients with solid tumors, the selection of target antigens is critical. The best candidate antigens, also called neoantigens, should be expressed only in malignant cells and not in non-malignant cells. These antigens may arise from tumor-specific nonsynonymous mutations, insertions, or deletions that alter the amino acid sequence of cell surface proteins, aberrant expression of cancer fetal antigens, or tumor-specific post-translational modifications. However, cell surface neoantigens are rare, especially in tumors with low mutational load. Therefore, most CAR T cell therapeutic targets in solid tumors are tumor-associated antigens (TAAs) also expressed in non-malignant tissues. Examples of TAAs include EGFR, HER2, CAIX, B7-H3, mesothelin, and GD2.
Preclinical Predictability of OTOT
The risk of OTOT in CAR-T cells has been studied in preclinical models. Histological analysis of mouse organs can detect the infiltration of CAR-T cells, the extent of tumor and normal tissue necrosis, and the expression levels of TAA around the tumor and at distant inflammatory sites. Immunohistochemistry can be used to verify the possibility of extra-tumoral TAA expression and CAR-T cell infiltration, providing clear evidence for OTOT. In addition, luciferase labeling of mouse CAR-T cells can also be used to monitor CAR-T cell trafficking and activation, thereby improving the understanding of CAR-T cell migration and extratumoral interactions.
Although mouse models can be used to study OTOT, differences in target antigen expression between mouse and human tissues also require attention when predicting clinical OTOT. For example, gastrointestinal OTOT responses occur in humans in patients treated with anti-CLDN18.2 CAR-T cells, but are not observed in mouse models. This is mainly due to differences in antigenic structure, target expression patterns, or different tissue microenvironments between human and mouse CLDN18.2 homologs.
The predictive power of mouse models can be enhanced using different approaches. This includes genetic engineering, replacing mouse genes with human homologs, or transplanting a cell line into mice to mimic TAA expression in extra-tumorigenic non-malignant tissues. However, these approaches still do not accurately reflect the complex expression patterns of TAAs in non-malignant human tissues. Modern organ-on-a-chip technology aims to mimic living microstructures and fully humanized tissue physiological processes, and while this approach may help address the shortcomings of traditional models, it still cannot fully mimic the complexity of antigen expression in human tissues.
Engineering Strategies to Mitigate OTOT
Toxic responses observed in CAR-T cell clinical trials have contributed to the development of techniques to improve safety while maintaining antitumor efficacy. New approaches are now emerging that use synthetic biology to better limit the potent cytotoxicity of CAR-T cells and thus avoid OTOT.
- Fine-tuning the CAR domain
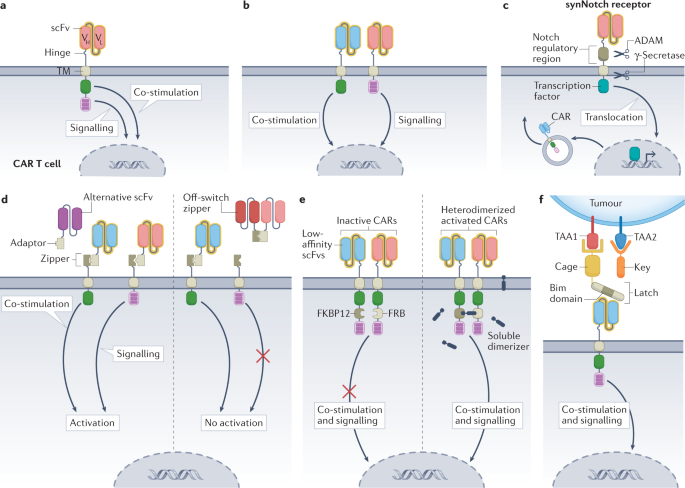
Affinity regulation by modifying the scFv of the CAR can achieve sufficient affinity for tumor cells without affecting non-malignant cells with limited TAA expression. This can be achieved by mutagenizing existing single-chain antibodies or by screening single-chain antibody libraries to identify alternative binding agents with different affinities. High affinity CAR-T cells may have better responsiveness to tumor cells with low antigen expression density, but may also lead to recognition of target antigens present in non-tumor tissues. Single-chain antibodies with very high affinity for target antigens may limit the potential for CAR-T cell proliferation. Conversely, low-affinity CAR-T cells may lack antitumor activity because they are unable to adequately recognize and lyse tumor cells with low levels of TAA expression. In addition, low-affinity single-chain Fv increases the risk that low-antigen density tumor cells will evade CAR-T cell recognition.
In addition to affinity regulation of scFv, modification of hinge and transmembrane structural domains (H/T) and quantitative regulation of immunoreceptor tyrosine-based activation motifs (ITAMs) can also alter the antigen density threshold of CAR-T. For example, 4-1BB-CD3ζ CAR-T cells with CD8-H/T have a higher antigen density threshold and form unstable immune synapses compared with CD28-H/T. In addition, reducing the number of ITAMs in the CD3ζ region can reduce the cytotoxicity of CAR-T cells against cells with limited antigen density while maintaining cytotoxicity against high antigen density targets. Loss-of-function mutations in one or more of the three ITAMs in CD3ζ can also be used to calibrate activation and differentiation programs, and the location of mutated ITAMs can determine CAR-T cell function, differentiation, and antitumor activity.
- Logic gate of CAR-T cells
Boolean logic gate methods, referring to the mathematical operators “IF-THEN”, “AND”, “OR”, and “NOT “, can be used to control the activation of CAR-T cells. Some of these methods can be used to increase the specificity of cell killing and reduce the likelihood of OTOT.
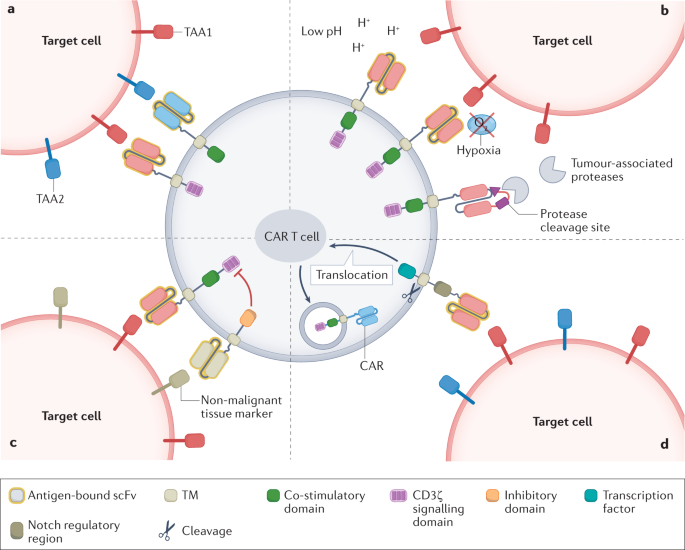
- Decoupling CAR-T cell activation signals using “AND” logic gate
Different CAR-T cell activation signals can be decoupled by “AND” logic gate to avoid OTOT in some cases. Dual “AND” logic CAR-T cells express two different synthetic receptors (one containing CD3ζ and the other containing CD28 or 4-1BB signaling domains), each targeting a different TAA in a split fashion. This dual control theoretically reduces the risk of OTOT, as both TAAs need to be expressed in non-malignant tissues for full CAR-T cell activation, and linking only one receptor results in attenuated signaling. The “AND” logic gate CAR-T cells targeting CEA and mesothelin exhibited specific cytotoxicity against mouse double TAA-positive cells, while not recognizing single TAA-positive cells. Another study showed more potent killing of double-TAA-positive cells using this approach. Such a strategy reduces the likelihood of OTOT, but “AND” logic gate relies on multiple TAAs, which increases the risk of antigen escape.
- Sensing TME helps construct “IF-THEN” logic gate
The “IF-THEN” logic gate provides an alternative strategy for limiting CAR-T cell killing of TMEs. pH-sensitive CAR-T cells optimally recognize TAAs in an acidic environment, providing an example of “IF-THEN” logic gate-based targeting. This approach takes advantage of the Warburg effect, which is prevalent in many tumors and refers to accelerated glycolysis and increased lactate production, resulting in an acidic intratumoral environment. HER2-targeting CARs with pH-restricted binding domains preferentially detect HER2 in acidic TME, a strategy that induces CAR-T cell expansion and regression of high antigen density HER2-positive tumor cells in mouse models.
Hypoxia induction also limits CAR expression and activity on TME. In one study, the hypoxia-sensitive sub-structural domain of HIF1α was fused to the C-terminus of a multistranded anti-CD19 CAR cell. Under normoxic conditions, the HIF1α structural domain and CAR cells were degraded. However, under hypoxic conditions at TME, HIF1α is not degraded and CAR can achieve local expression.
Another approach uses protease-sensitive linkers to attach the shielding peptide to the antigen-binding site of the anti-EGFR CAR. The antigen-binding site of the CAR can only be released by protease cleavage of the shielding peptide. This strategy limits the function of the CAR to the TME, which has locally high concentrations of membrane-type serine protease 1 (MT-SP1), urokinase-type fibrinogen activator (uPA), and lysosomal enzymes. Such protease-dependent CARs provide levels of antitumor activity comparable to those observed with unmodified CARs, but with greater selectivity. These approaches also have shortcomings, for example, certain non-malignant tissues may share the targeting profile of TME, leading to activation of TME-sensitive CAR-T cells at locations far from the tumor. Therefore, for functional T cell therapy with hypoxia or pH-induced CAR expression, the selection of target antigens should focus on TAAs that are not expressed in non-malignant hypoxic and acidic tissues.
- SynNotch circuits: use “IF-THEN” logic gate to promote multiple antigen sensing
CAR-T cells that use “IF-THEN” logic gate of synthetic Notch (SynNotch) circuits rely on multiple antigen sensing, overcoming the trade-off between affinity and selectivity. By limiting CAR-T cell effects to multiple tumor targets, this approach allows the selection of more aggressive, higher affinity CARs that might otherwise lead to OTOT. SynNotch CAR-T cells are activated in two steps, and CAR-T transgene expression is induced only after the initiating antigen is recognized by the SynNotch receptor. In the initial report of SynNotch CAR-T cells, CAR-T cell expression and T cell activation depended on the recognition of two different TAAs. SynNotch CAR-T cells were able to clear double TAA-positive tumors, while cells expressing one TAA survived.
The SynNotch circuit is versatile, allowing the use of multiple SynNotch receptors to be selected for designing more specific and less toxic CAR-T cells. However, the potential for host immune response to SynNotch proteins and the complexity of transgenesis are the greatest challenges to the clinical application of this technology. To overcome these difficulties, researchers have redesigned the underlying SynNotch structure and created a system of “SyNthetic intramembrane proteolysis receptors” (SNIPRs) that are more compact and compatible with human transcription factors. However, the migration of initiated SynNotch CAR-T cells from tumors to non-malignant tissues expressing CAR antigens remains a potential risk.
- Versatile CAR design with “AND” logic gate allows exogenous control
Synthetic biology has built more complex logic systems that include modifiability of cellular activity after infusion of CAR-T. For example, a discrete, universal and programmable (called SUPRA) CAR can be modified to combine different logic gate strategies. A functionally complete SUPRA CAR contains two elements: (1) a transmembrane inner domain of the CAR attached to the extracellular leucine zipper; (2) a soluble scFv attached to the leucine adapter (adapter-scFv). SUPRA CAR-T cells are activated only when adapter-scFv is bound to TAA and the adapter is attached to the CAR via the zipper. To avoid OTOT, the SUPRA circuit can be further programmed. In this case, two SUPRA CARs can be designed so that the CD3ζ signal and the co-stimulatory signal are delivered by two independent receptors for antitumor activity. Furthermore, to prevent CAR-T cell signaling, soluble zipper-scFv competitive binding adapters can be used to block antigen recognition and CAR-T cell activation. In addition, an avidity-controlled CAR (AvidCAR) system is designed based on the concept of cellular affinity, which activates only when two low-affinity CARs independently recognize target antigens and dimerize upon induction of specific small molecule dimerization agents. This design can be modified to combine CARs with scFvs of different TAAs, thus turning the system into a proximity-dependent and -logic gate.
Other Strategies to Mitigate OTOT
- CAR-T-cell infusion followed by clinically approved drugs to control OTOT
Systemic immunosuppression with corticosteroids is currently the strategy of choice to manage most immunotherapy-related toxicities. However, administration of corticosteroids may negatively impact the antitumor effect and persistence of CAR-T cells. Reversible (or temporary) control of CAR-T cells can be achieved with the tyrosine kinase inhibitor dasatinib, which acts as an immediate off switch for CAR-T cell signaling without affecting cell viability.
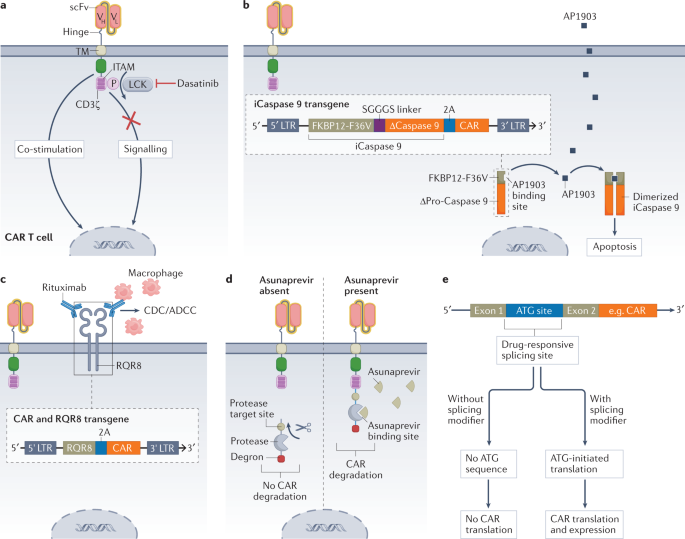
- Control of OTOT by a suicide switch
Suicide switches and other strategies that can eliminate or inactivate CAR-T cells are important safety measures for the treatment of life-threatening adverse events. Various strategies have been tested, including the use of an inducible Caspase 9 (iCaspase 9). This construct can be activated by the small molecule AP1903 to induce apoptosis of CAR-T cells within minutes of exposure. Data from a clinical study published in 2021 suggest that iCaspase 9 can be used to safely ameliorate clinically severe neurotoxicity in patients receiving anti-CD19 CAR-T cell therapy.
- Controlling OTOT by modulating CAR expression and cytotoxicity
Irreversible elimination of CAR-T cells using a suicide switch prior to complete tumor eradication may limit clinical efficacy. Another option may be to design reversible switches that allow controlled CAR-T cell activation for a certain period of time and maintain anti-tumor function. In a switch system, CAR surface expression could be controlled using the protease inhibitor asunaprevir. To achieve this effect, a protease target site, a protease, and a degron are fused to the C-terminus of the CAR. In the absence of asunaprevir, the protease separates the degron from the CAR structure, thus maintaining CAR surface expression. After administration, the protease is inhibited and degron cannot be cleaved, which leads to CAR degradation. Systems for CAR binding to ligand-induced degradation domains have been developed, and this approach allows CAR degradation by the proteasome in the presence of the ligand, resulting in adjustable control of CAR expression in vivo.
- Control of CAR expression by acousto-optics
Ultrasonography can be used to control the transcription and expression of CAR. Ultrasound-sensitive piezoelectric ion channels can trigger signal cascades. In August 2021, researchers discovered a method to place the expression of anti-PSMA or anti-CD19 CARs under the regulatory control of heat shock proteins. This approach allows controlled expression of CAR in vivo by applying focused ultrasound, which raises the temperature inside the tumor to approximately 43°C, thus inducing a tumor-limited cytotoxic response while limiting cytotoxicity outside the tumor.
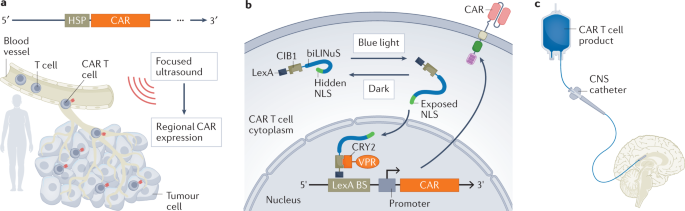
- CAR-T cell local delivery
Traditionally, CAR-T cell delivery has been by systemic injection, which poses a challenge for adequate CAR-T cell transport to the tumor site and minimal transport to non-malignant tissues. Local delivery and targeted transport can help overcome these challenges. For example, CAR-T cells targeting CNS tumors have shown better preclinical and early clinical outcomes when administered intracerebroventricularly and intrathecally. Intracranial infusion of anti-il13r α2 CAR-T cells resulted in transient anti-tumor responses in 2/3 of patients with glioblastoma. Preliminary data from this trial suggest that repeated local administration is well tolerated and elicits a local immune response. In March 2022, researchers found successful intracerebroventricular administration of anti-GD2 CAR-T cells in patients with diffuse intrinsic pontine glioma. 3/4 of patients had clinical and imaging improvement, and although GD2 was also expressed in normal brain tissue, no OTOT response was observed.
Summary and Outlook
CAR-T cell therapies have been modified to greatly enhance their efficacy in treating solid tumors and are steadily progressing toward clinical trials. The identification of a large number of TAAs has laid the foundation for the use of CAR-T cell therapy in patients with solid tumors, although their expression in non-malignant tissues may predict an increased risk of OTOT. However, current strategies to assess OTOT risk are imperfect, and clinical OTOT responses are often inconsistent with preclinical data. Therefore, there is a need to refine animal models and advance in vitro systems in future studies to more accurately predict OTOT.
A significant amount of research is now focused on developing more specific CAR systems, including systems that allow exogenous control of T cell function or survival. Adjusting the affinity of scFvs and changing CAR architecture may reduce the risk of OTOT. Various protein-based logic gate strategies, which may also further limit CAR-T cell activation and tumor-killing sites, have also been explored in preclinical models. Other engineering approaches that control CAR-T cell activity exogenously or eliminate CAR-T cells in a timely manner can also reduce the risk of OTOT. In addition, local administration of CAR-T cells in the tumor environment may improve antitumor activity while avoiding OTOT responses to a greater extent. In addition to these approaches, developing novel CAR-T cells for solid tumors and addressing the risk of OTOT are critical for the clinical safety of drug use.
Reference
1. Flugel, Christian L., et al. “Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours.” Nature Reviews Clinical Oncology 20.1 (2023): 49-62.