In 2013, experts reached a consensus to adopt the term C3 glomerulopathy (C3G) to define a group of rare kidney diseases driven by dysregulation of the alternative complement pathway, mediated by genetic mutations or acquired defects in the complement cascade. Its histopathology is characterized by the accumulation of complement C3 in renal tissue. It is mainly divided into two subgroups: Dense Deposit Disease (DDD) and C3 Glomerulonephritis (C3GN).
C3G disease is not a newly recognized entity. DDD was formerly known as MPGN Type 2, and C3GN was previously classified as atypical MPGN type 1 or 3. To better understand the role of complement in pathogenesis, they have been reclassified into a different group of diseases, C3 glomerulopathy.
Incidence
The rarity of C3 glomerulopathy makes it difficult to obtain accurate incidence and prevalence data. However, estimates with limited reliability are available from some small cohort studies. In the United States, the incidence of C3 glomerulopathy is estimated to be 1-3/1,000,000 based on an analysis of data from the C3 glomerulopathy registry. Estimates from four European studies are 0.2-1 in 1,000,000.
Pathogenesis
Complement system
The complement system consists of more than 50 individual proteins or activation fragments.
The complement system is essential for both innate and adaptive immunity: a delicate balance between activation and regulatory mechanisms enables the system to target infectious microorganisms, clear immune complexes and apoptotic cells, and enhance humoral responses.
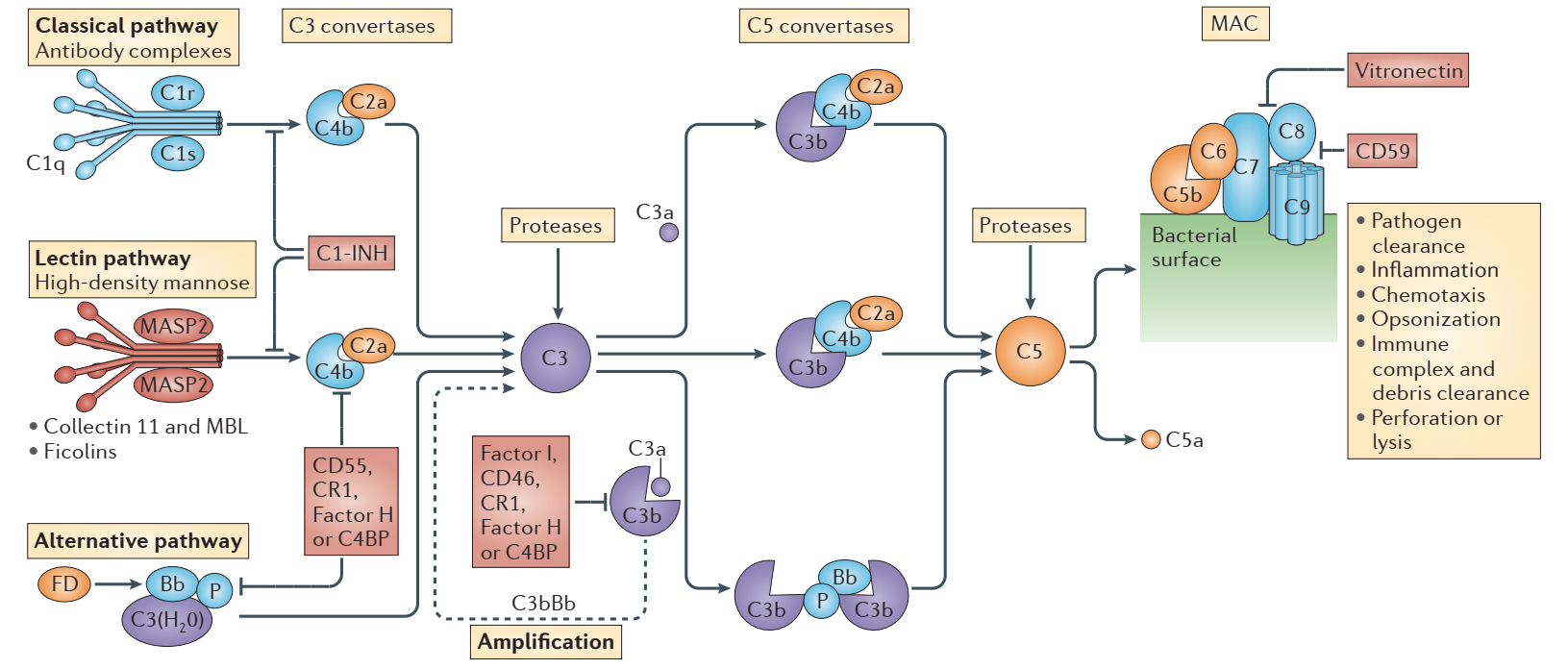
Complement is activated by classical, lectin, and alternative pathways. Each pathway has a unique trigger. Activation of the classical pathway involves the recognition of antigen-antibody complexes. The lectin pathway is triggered by microbial polysaccharides. Initiation of the alternative pathway depends on natural hydrolysis of C3.
Once activated, these pathways lead to the formation of two C3 convertases, C4b2a via canonical and lectin pathways, and C3bBb via the alternative pathway.
C3 convertase cleaves C3 into C3a and C3b, and C3b docks with factor B, which is cleaved by factor D to generate additional C3bBb.
As more C3bBb is formed, the terminal pathway is activated by C3bBbC3b, and a small amount of C4b2aC3b, the C5 convertase produced by the classical and lectin pathways. C5 convertase cleaves C5 into C5a (a potent anaphylatoxin) and C5b (initiates the terminal pathway), ultimately producing soluble C5b-9 or membrane attack complex (MAC), which induces cell lysis.
C3 is the acute phase reactant. It is produced in large quantities by the liver. As one of the most abundant plasma proteins, C3 circulates at concentrations of ~1.2mg/ml, ensuring its immediate response to stimuli, such as the presence of infectious microorganisms.
Pathogenesis of C3 glomerulopathy
Dysregulation of the alternative complement pathway underlies C3 glomerulopathy, and the impact of complement dysregulation manifests itself in the glomerular microenvironment.
Animal models are valuable for demonstrating that uncontrolled activation of the alternative pathway drives the pathogenesis of C3 glomerulopathy. It was initially demonstrated in a pig model of DDD. Then in mouse studies, it was found that the absence of complement factor H (Cfh-/- mice) led to spontaneous development of kidney injury and renal pathology similar to human C3 glomerulopathy. In the absence of complement factor H and factor B (Cfh-/-Cfb-/- mice), C3 convertase (C3bBb) cannot be formed, and kidney disease does not develop. If C5 is absent (C5-/- mice), C3 glomerulopathy cannot be prevented despite the lack of terminal pathways, but disease severity is significantly reduced.
According to statistics, approximately 25% of patients with C3 glomerulopathy have genetic variants and defects.
Variant and defective genes:
- Genes for two invertases: C3 and factor B.
- Regulatory protein genes: H factor and I factor.
- Complement activation enhancer: After gene mutation and recombination of complement H-related protein (CFHR), a new fusion gene is generated, which is transcribed and translated into a new FHR fusion protein.
It is not unusual for patients with C3 glomerulopathy to carry multiple variants in complement-related genes. Such genetic complexity may partly explain why families with first- or second-degree relatives of an affected individual with the same diagnosis are very rare.
About 75% of C3 glomerulopathy results from acquired factors. Autoantibodies against various complement proteins cause complement chain dysregulation.
- C3 nephritic factor: present in up to 80% of DDD and 50% of C3GN patients.
- C5 nephritic factor: more frequently detected in C3GN patients than in DDD patients.
- C4 nephritic factor.
- Autoantibodies against factors H and B.
Clinical manifestations and outcomes
C3 glomerulopathy has a wide range of clinical manifestations, ranging from asymptomatic hematuria and proteinuria to acute presentation with typical signs and symptoms of glomerulonephritis. Patients usually have a history of severe hematuria and hypertension and may also have an associated history of acute kidney injury (AKI) and/or chronic kidney disease (CKD). Most patients have low serum C3 levels.
All age groups are affected, with DDD having a lower mean age than C3GN.
C3 glomerulopathy will be clinically transformed into ESRD. 70% of children and 30-50% of adults develop ESRD within 10 years of diagnosis.
Renal pathology
C3 glomerulopathy is a histopathological diagnosis. Itis defined by the presence of unique (or at least significant) positivity for C3 by immunofluorescence staining at least two orders of magnitude higher in intensity than any other immunofluorescence in renal biopsy samples.
Light microscopy findings are varied and range from no glomerular hyperplasia to mesangial hyperplasia, intracapillary hyperplasia, exudation, membranous hyperplasia, crescents, and sclerosis.
Electron microscopy distinguishes two major subtypes of C3 glomerulopathy:
- DDD: Electron microscopy shows high electron density, osmium-loving deposits with a “sausage-like” appearance that thickens and deforms the dense layer of the glomerular basement membrane. Similar ultradense deposits can also be identified in Bowman’s capsule and some tubule basement membranes.
- C3GN: The electron density of the sediment is close to that of the glomerular matrix component. These deposits typically have an amorphous, cloudy appearance within the mesangium and may appear as poorly defined subendothelial (intramembranous and/or subepithelial) inclusions.
- Subepithelial hump occurs in both subtypes.
Differential diagnosis
Immune complex glomerulonephritis (ICGN): It depends on the immunostaining technique and tissue preparation used. Immunofluorescence on frozen tissue is more sensitive and reliable than immunoperoxidase on formalin-fixed, paraffin-embedded tissue for grading the intensity of C3 staining. Additionally, some of the media can mask any immunoglobulins present, preventing them from being detected by routine tests.
Post-infectious glomerulonephritis (PIGN): If the co-deposition of IgG and C3 and predominant glomerular deposition of C3 are observed, it usually represents the advanced stage of the disease. The 2013 consensus meeting acknowledged the similarities of PIGN and C3 glomerulopathy and the difficulty in distinguishing the two diseases based on pathological features alone. The patient’s clinical course and laboratory findings are ultimately differentiated. Renal function returned to baseline in almost all patients with PIGN without therapeutic intervention within a few weeks, with improvements in hematuria, proteinuria, and hypocomplementemia.
Disclaimer: Creative Biolabs focuses on promoting biological and biomedical research globally. This article is for information exchange purposes only. This article is also not a treatment plan recommendation. For guidance on treatment options, please visit a regular hospital.