Creative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsWith over a decade of experience in phage display technology, Creative Biolabs can provide a series of antibody or peptide libraries that are available for licensing or direct screening. These ready-to-use libraries are invaluable resources for isolating target-specific binders for various research, diagnostic or therapeutic applications.
HighlightsCreative Biolabs has established a broad range of platforms for developing novel antibodies or equivalents. These cutting-edge technologies enable our scientists to meet your demands from different aspects and tailor the most appropriate solution that contributes to the success of your projects.
HighlightsWith deep understanding in antibody-related realms and extensive project experience, Creative Biolabs offers a variety of references to help you learn more about our capacities and achievements, including infographic, flyer, case study, peer-reviewed publications, and all kinds of knowledge that can assist your projects. You are also welcome to contact us directly for more specific solutions.
HighlightsGet a real taste of Creative Biolabs, one of the most professional custom service providers in the world. We are committed to providing highly customized comprehensive solutions with the best quality to advance your projects.
HighlightsMetabolism is one of the key parameters to be investigated and optimized in drug discovery projects. In silico approaches to address metabolic events cover a number of different aspects, ranging from the prediction of the overall metabolic stability of a compound or the strength of binding to metabolic enzymes to the calculation of microscopic reaction barriers. Creative Biolabs is dedicated to establishing the most exquisite service platform to provide one-stop computational pharmacology services for global clients.
For about 75% of all drugs, metabolism is one of the major clearance pathways. The process of biotransformation can produce metabolites with substantially altered physicochemical, physiological, pharmacological, and toxicological profiles. Metabolic systems are highly complex and adaptable. A plethora of diverse enzyme families is involved in xenobiotic metabolism, including cytochrome P450 (CYP) enzymes, dehydrogenases, flavin-containing monooxygenases, hydrolases, peroxidases, UDP-glucuronosyltransferases (UGTs), sulfotransferases, and glutathione S-transferases. The identification of metabolites is a key task, as they may result in reactive species, toxic, or even active compounds, and ideally, the modification of the compound to improve the pharmacokinetic profile by changing the metabolic susceptibility is an important step. Consequently, computational methods for predicting the possible site of metabolism of compounds are extremely useful.
According to the strategies used for prediction, in silico methods can be broadly classified into two categories: ligand-based approaches, which use the information of the substrate (ligand); and structure-based approaches, which use the information of the enzyme-substrate complex.
Ligand-based approaches rely on the assumption that the metabolic fate of a compound is exclusively a consequence of its chemical structure and characteristics. These methods depend on a set of known substrates or inhibitors to build models, providing indirect information about the enzyme’s active site. The shape, electronic properties, and conformations of these small molecules are taken into consideration, assuming that such characteristics will determine the compound’s metabolic pathway. All ligand-based approaches need to handle uncertainties about the binding cavity of a protein. However, small changes in the structure of a xenobiotic do not necessarily result in major changes in its metabolism, presumably due to the high flexibility and plasticity of the metabolizing enzyme, such as P450s.
Structure-based models not only include properties of the substrate but also explicitly model the interaction between the substrate and the enzyme in question. Drug metabolism studies allow the prediction of the binding modes of substrates, the conformational change of enzymes, the mechanism of catalysis, and the elucidation of metabolic differences in enantiomers. To perform structure-based metabolism prediction, structures of P450s or other drug-metabolizing enzymes are required and can be experimentally determined using X-ray crystallography, nuclear magnetic resonance (NMR), or electron microscopy. Obtaining a high-resolution 3D structure can be challenging for many proteins and this obstacle led to the creation of theoretical homology models. Currently, with the remarkable advances in the proteomic area, several human P450 structures became available in the RCSB Protein Data Bank (PDB).
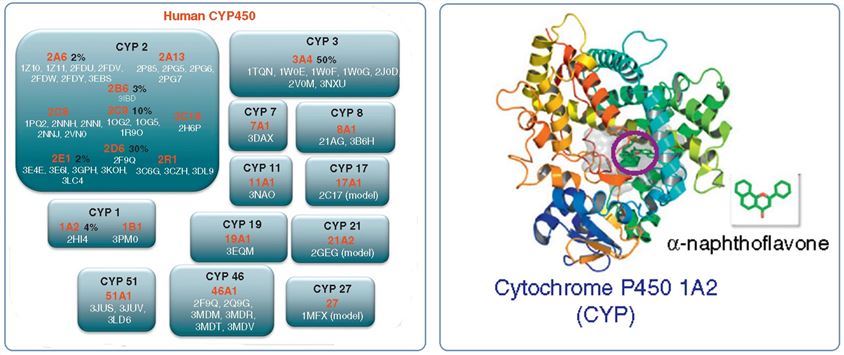 Fig.1 Available 3D structures from the PDB for CYPs.
Fig.1 Available 3D structures from the PDB for CYPs.
In silico prediction of drug metabolism has advanced greatly in the past few years, and most properties can now be predicted. Prediction of physicochemical properties is in some cases as good as experimental measurement. Nevertheless, predicted drug metabolism is increasingly used in lieu of measured values, and this trend is expected to continue as predictions become more accurate, and systems are designed to handle the multi-factorial nature of metabolism contributions to drug development. This will have the effect of shortening the development time and hence reducing the cost of new drugs by alleviating or even eliminating the metabolism bottleneck. We can provide a variety of computational methods for predicting drug metabolism and toxicity modeling to meet customers’ specific requirements.
Creative Biolabs has focused on the development of computational drug design for years, and we whole-heartedly cooperate with you to accomplish our shared goals. Our team provides you with outstanding support and meets your specific needs with a professional technology platform. If you are interested in our services, please contact us for more details.
All listed services and products are For Research Use Only. Do Not use in any diagnostic or therapeutic applications.