ATP1A3 and Associated Diseases
With rich experience for over a decade, Creative Biolabs manifests as a first-class service provider in the field of gene therapy development. We integrate global resources to provide not only a full range of gene therapy products and testing services but also comprehensive support knowledge about the ATP1A3 gene and associated diseases. Our experts are willing to help you to understand the molecular pathogenesis involved in ATP1A3-associated diseases and greatly assist in your research and development.
Background of ATP1A3
The ATP1A3 gene on chromosome 19q encodes the α3 isoform of Na+/K+-ATPase, which is required as a rescue pump. The α3 isoform plays a role in the rapid restoration of large transient increases in intracellular Na+ levels after repeated action potentials. ATP1A3 is expressed almost exclusively in all neurons of the central nervous system (CNS), coexisting with neurons that produce gamma-Aminobutyric acid (GABA).
Na+/K+-ATPase consists of a heterogeneous α-β-γ protein complex organized into multiple isoforms, which is a transmembrane sodium-potassium pump at the cell plasma membrane involved in controlling the sodium and potassium ion concentration gradients. For every 1 ATP consumption, the Na+/K+-ATPase pumps 3 Na+ ions out of the cell and 2 K+ ions into the cell. The normal function of the Na+/K+-ATPase plays an important role in osmotic regulation, the electrical excitability of nerves and muscles, as well as sodium-coupled transport.
ATP1A3 Associated Diseases
The heterozygous pathogenic variants of the ATP1A3 form a large number of phenotypic variants of the α3 isoform, which cause the dysfunction of Na+/K+-ATPase. The dysfunction of Na+/K+-ATPase due to the alternatively spliced transcript variants of the ATP1A3 gene encoding different isoforms leads to a range of inherited neurological syndromes, including rapid-onset dystonia parkinsonism (RDP), cerebellar ataxia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS syndrome), and alternating hemiplegia of childhood (AHC).
- RDP
RDP is an autosomal dominant disorder with an abrupt onset. Usual syndromes of RDP include craniocervical dystonia, mild limb dystonia, parkinsonism with no pill-rolling tremor, bulbar symptoms including general dysarthria and hypophonia with dysphagia, and cognitive impairment. The p.Thr613Met of the ATP1A3 gene is known to be the most frequent pathogenic variant causing RDP.
- AHC
AHC is a syndrome mostly occurring in infancy, which presents with developmental delay, episodes of intermittent hemiplegia on alternating sides of the body, choreoathetosis, and dystonia. The variants of ATP1A3 in AHC occur in an amino acid. The p.Asp801Asn, p.Glu815Lys, and p.Gly947Args are the most common ATP1A3 pathogenic variants in AHC.
- CAPOS syndrome
CAPOS syndrome is a dominantly inherited syndrome defined by cerebellar ataxia with a relapsing course, sensorineural hearing loss, optic atrophy, areflexia, and pes cavus. The p.Glu818Lys is a pathogenic variant of ATP1A3 that leads to reduced Na+ affinity and accumulated Na+ associated with action potentials from neurons, which cause a range of axonal neuropathy.
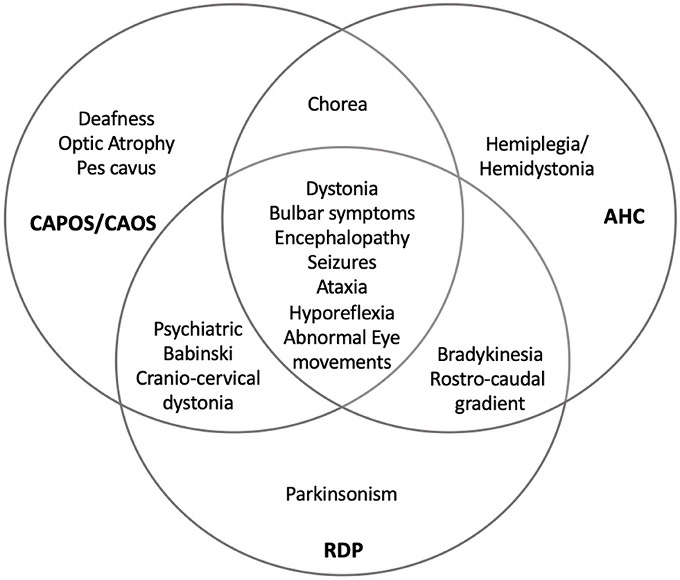 Fig.1 Overlapping of ATP1A3-related disorders. (Salles, 2021)
Fig.1 Overlapping of ATP1A3-related disorders. (Salles, 2021)
Many diseases are associated with ATP1A3 mutations. Comprehending the pathogenesis of ATP1A3-related diseases is the future focus of developing gene therapies. As a frontier biotech service provider, Creative Biolabs is dedicated to providing remarkable products and services in the gene therapy field. Our team offers global researchers advanced information about ATP1A3 and associated diseases. If you want to learn more support knowledge about the ATP1A3 gene and associated diseases, please don’t hesitate to contact us for more information.
Reference
- Salles, P. A. et al. ATP1A3-related disorders: an ever-expanding clinical spectrum. Frontiers in neurology. 2021, 12: 637890. Distributed under Open Access license CC BY 4.0, without modification.