Mechanisms of Oncolytic Virus Targeting Tumor Cells
Oncolytic viruses (OVs) are viral strains that can infect and kill malignant cells (oncolysis) while sparing their normal counterparts. Some viruses demonstrate a natural ability to target cancer cells selectively and more efficiently than normal cells. Cancer cells have several distinct hallmarks that separate them from normal cells: sustained growth signals, insensitivity to anti-growth signals, evasion of apoptosis, increased angiogenesis, cell immortality, and invasion/metastasis. OVs utilize several mechanisms to enter cancer cells,Creative Biolabs has summarized those regulations as following.
-
Abnormal cell receptors
Cancer cells have been shown to overexpress selected surface receptors, by which viruses may selectively bind to and infect cancer cells. -
Aberrant signaling pathways
In fact, some viruses naturally exploit the aberrant signaling pathways that maintain sustained cancer growth in order to selectively infect and replicate within cancer cells as opposed to normal cells. -
The hypoxic environment
The rapid proliferation of tumor cells results in the majority of solid tumor tissues in a hypoxic state. Fortunately, some oncolytic viruses can selectively infect tumor tissues in a low oxygen environment.
Abnormal cell receptors
Some viruses can enter cells through one or more receptors and some of which can promote the entry of more than one type of virus. Some viruses, like Newcastle disease virus (NDV), vaccinia virus (VV), and vesicular stomatitis virus (VSV), use endocytosis through membrane fusion and syncytia formation to enter cells. Certain oncolytic viruses, such as Seneca valley virus and reovirus, are known to preferentially target cancer cells but the cell surface receptor for entry has not been identified. Many of the oncolytic viruses currently in the clinic have a natural tropism for cell surface proteins aberrantly expressed by cancer cells.
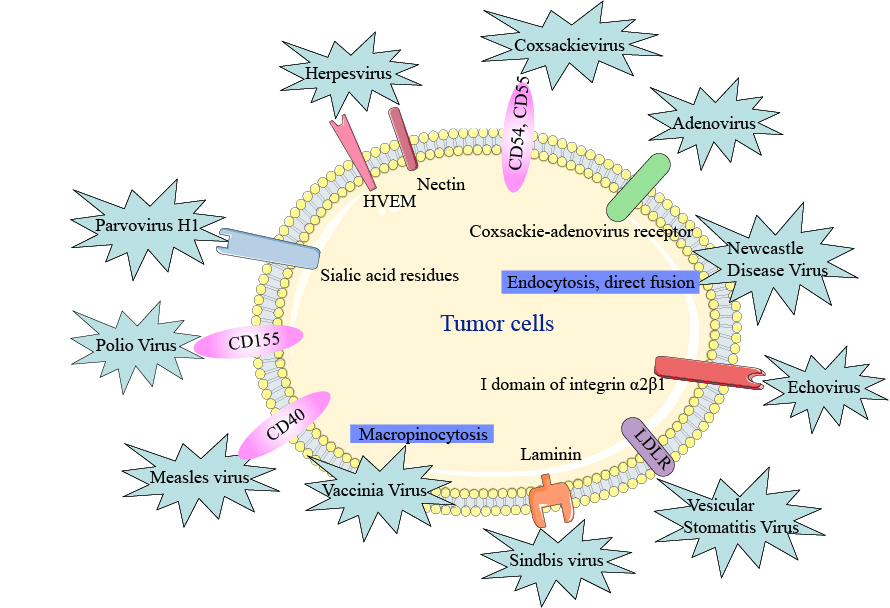 Figure 1 The tumor tropism of oncolytic viruses
Figure 1 The tumor tropism of oncolytic viruses
- Herpesvirus (HSV)
HSV completes cell entry through the herpesvirus entry mediator (HVEM) and selected nectins. These surface receptors are overexpressed on some cancer cells, including melanoma and various carcinomas.
- Coxsackievirus
Coxsackievirus can enter cells via intercellular adhesion molecule 1 (ICAM 1; also known as CD54) and decay-accelerating factor (DAF; also known as CD55), which can be overexpressed in cancers such as multiple myeloma, melanoma, and breast cancer (although coxsackieviruses may also use nectins for cell entry).
- Echovirus
Echovirus, a member of the enterovirus family, has increased specificity for ovarian cancer cells by using the I domain of integrin α2β1 for cell entry, which can be overexpressed on these cells.
- Sindbis virus
Sindbis virus, a member of the Togaviridae family, targets cancer cells that overexpress the 67kDa laminin receptor that promotes cancer cell invasion and motility.
- Measles virus
Measles virus, specifically the Edmonston strain, enters the cell by surface receptor CD46 that prevents cell elimination by inactivating the complement pathway of the immune system and is often overexpressed by tumor cells.
- Poliovirus
Poliovirus, another member of the enterovirus family, has enhanced specificity for various cancers through its targeting of CD155, a receptor that can potentially impair antitumor NK cell responses and is overexpressed by some tumor cells.
Aberrant signaling pathways
Oncolytic viruses can also target oncogenic pathways which are different from the expression in normal tissues, thus maintaining sustained cancer growth.
Regulation of cell cycle entry and proliferation in healthy cells is provided by key factors, such as protein kinase R (PKR), p16, retinoblastoma (Rb), and the tumor suppressor p53. These elements promote abortive apoptosis when the cell cycle is dysregulated. PKR may also help to regulate transcription and induce abortive apoptosis when cells are infected with a virus. The expression of oncogenes and other aberrant host cell proteins in cancer cells can promote viral replication and oncolytic activity.
In cancer cells, cell cycle regulation and cellular proliferation are typically disrupted due to the activity of oncogenes and the loss of tumor suppressor genes. These changes can support viral replication and promote oncolytic virus-induced cell death.
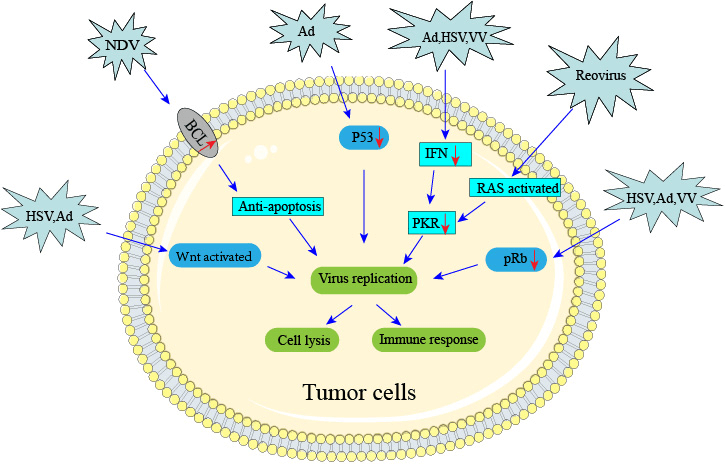 Figure 2 Mechanisms of viral targeting aberrant signaling pathways
Figure 2 Mechanisms of viral targeting aberrant signaling pathways
- Targeting defective p53 pathway
The p53 tumor suppressor pathway is inactivated in nearly all human tumors either through direct mutation of p53 or the loss of upstream regulators such as p14ARF or downstream p53 effectors such as Bax. DNA damage or activated oncogenes induce p53, which activates the transcription of downstream p53 effectors, resulting in the cell cycle arrest or apoptosis of cells that may have acquired potentially tumorigenic lesions. Thus, inactivation of the p53 tumor suppressor pathway is a critical event for tumorigenesis. Some viruses, including adenovirus, reovirus and parvovirus, preferentially target p53 mutant or p53-null cancer cells because healthy cells with intact p53 undergo abortive apoptosis upon infection. Like cancer cells, DNA viruses inactivate the p53 pathway, and many encode proteins that bind and degrade p53. E1B-55K, a gene product of mutant adenovirus, appears to work together with E4-ORF6 and cellular proteins involved in ubiquitination and induces the degradation of p53. ONYX-015 (dl1520) is an adenovirus mutant that lacks the E1B-55K gene and, therefore, fails to degrade p53 during viral replication. On this basis, ONYX-015 is restricted for replication in normal cells, but is capable of replicating in p53-deficient tumor cells.
- Targeting Wnt signaling pathway
Aberrant activation of the Wnt pathway is implicated in driving the formation of various human cancers, particularly those of the digestive tract. Activation of the pathway results mainly from mutations in the adenomatous polyposis coli (APC) and β-catenin genes. In response to wnt signals, GSK3β is inhibited and β-catenin is stabilized. β-catenin then enters the nucleus, binds to Tcf/Lef family transcription factors, and activates transcription of wnt target genes, such as cyclin D, myc, and PPARδ. Taking advantages of this unique feature, CRAds was developed to express the viral E1B and E2 genes from promoters controlled by the Tcf4 transcription factor. Additional improved versions of oncolytic Ads targeting the wnt pathway have been made and tested in tumor models with defective wnt signaling. Based on the same principles, a replicating parvovirus was also developed. However, the virus replicated poorly in wnt activated cancer cells and further improvements remained challenging. In contrast, an oncolytic HSV (bM24-TE) constructed with the ICP4 gene, of which the viral replication is driven by a synthetic promoter containing the tandem repeats of a Tcf responsive element, is efficacious against colorectal cancers carrying an APC gene mutation between the first and second 20-amino-acid repeats.
- Targeting defective IFN, EGFR/Ras, PRK signaling pathways
When infected by a virus, cells are stimulated to produce IFNs that will activate an antiviral defense response of nearby cells. When IFNs bind to cell surface receptors (IFN-R) on neighboring cells, an intercellular signal transduction cascade is engaged to induce PKR expression. The kinase PKR is an important downstream effector molecule of the IFN signaling pathway. The dsRNA produced by viruses will bind to PKR, and lead to autophosphorylation of the PKR homodimer and activation of its kinase activity. The activated PKR phosphorylates eIF-2α, which in turn inhibits protein synthesis and thereby viral replication in the cells. Ad, HSV and VV encode a number of viral proteins to inhibit the antiviral response. For examples, virus-associated (VA) RNAs produced by Ad, E3L and K3L proteins produced by VV and NS1 protein produced by IFA all can inactivate PKR. Cancer cells have accrued many mutations such as inactivating mutations in these signaling pathways. Oncolytic VSV targets this characteristic for its natural oncotropism. A constitutively activated Ras signaling pathway, another common feature for tumor cells, leads to the inhibition of PKR autophosphorylation. Oncolytic NDV and reovirus depend on this characteristic for their tumor cell selectivity. Both down-regulated IFNs and activated Ras in tumor cells can block the activation of PKR, and make the virus successfully replicate in tumor cells.
- Targeting anti-apoptosis pathways
Many cancer cells get involved in evading apoptosis. Apoptosis is characterized by certain morphological and biochemical changes that are well described. The B-cell lymphoma 2 (Bcl-2) family of proteins is one of the most extensively studied regulators of apoptosis. Therapeutically, apoptosis is important as many antineoplastic agents cause cellular damage by activating the apoptosis pathway. Resistance to these agents has attributed to the overexpression of antiapoptotic proteins of the Bcl-2 family, especially Bcl-xL, an antiapoptotic protein that in some cancers has been shown to confer resistance to antineoplastic agents. During evolution of host pathogen interactions, viruses have also evolved mechanisms to evade apoptosis in order to assist their replication. The viral genes encoding anti-apoptosis functions are therefore theoretically expendable in cancer cells but essential for viral replication in normal cells. Research indicated that, deletion of multiple viral genes has generated tumor-selective and potent OVs in oncolytic viruses including Ad, HSV and VV.
- Targeting pRb pathways
The retinoblastoma tumor suppressor protein (pRb) pathway is dysregulated in a majority of human cancers and is targeted by the Conserved Region 2 (CR2) of the E1A protein. As an example, the E1A gene of mutant Ad is modified to target the Rb pathway. Moreover, the human E2F-1 promoter is selectively activated in tumor cells with a defect in the Rb pathway. Therefore, an alternative means to achieve tumor selectivity in Ad is to control an essential viral gene with an E2F-1 promoter to target Rb pathwaydefective tumor. Aberrant expression of Rb and p16, which regulates cell cycle entry, can render cancer cells susceptible to oncolytic viruses such as adenovirus, HSV-1, vaccinia virus and reovirus.
Target the hypoxic environment
Tumor hypoxia presents an obstacle to most antitumor therapies, including treatment of oncolytic viruses. That is, an oncolytic virus must resist the inhibition of DNA, RNA, and protein synthesis that occurs during hypoxic stress.
The vesicular stomatitis virus (VSV), an oncolytic RNA virus, is capable of replication under hypoxic conditions. VSV infection under hypoxic stress produces larger amounts of mRNA than under normoxic conditions. However, translation of these mRNAs was reduced at the early stage of postinfection in hypoxia-adapted cells than in normoxic cells. At the late stage of postinfection, VSV overcame a hypoxia-associated increase in subunit of eukaryotic initiation factor 2 (eIF-2) phosphorylation and initial suppression of viral protein synthesis in hypoxic cells to produce large amounts of viral protein. VSV infection causes the dephosphorylation of the translation initiation factor eIF-4E and inhibites host translation similarly under both normoxic and hypoxic conditions. VSV produces progeny virus to similar levels in hypoxic and normoxic cells, showing the ability to expand from an initial infection of 1% of hypoxic cells to an entire population. In general, VSV has an inherent capacity for infecting and killing hypoxic cancer cells.
Based on these different targeting mechanisms, engineered viruses with optimized targeting capabilities have been continuously developed, enabling oncolytic viruses to target cancer cells more efficiently and accurately.
Creative Biolabs is a company with a deep knowledge background, we have provided you with more detailed message about inherent and engineered oncolytic viruses. Besides, taking advantage of our oncolytic virus engineering platform, Creative Biolabs also provides you with comprehensive target site information and modification methods.