CFH Structure CFH Functions CFH Test CFH Deficiency Therapeutic Target
Complement Factor H (CFH) is the cornerstone of the complement system and a key regulator of immune homeostasis. This soluble glycoprotein protects host cells from collateral damage while enabling targeted elimination of pathogens. As a key player in the alternative complement pathway, dysregulation of CFH has been associated with a variety of conditions ranging from autoimmune diseases to age-related macular degeneration (AMD).
Creative Biolabs provides insights into the molecular structure, functional mechanisms, disease associations, and therapeutic potential of CFH, positioning it as a focal point for cutting-edge immunology research and drug development.
Structure of Complement Factor H
CFH is a 155-kDa plasma protein composed of 20 short consensus repeats (SCRs), each stabilized by disulfide bonds. These domains mediate distinct functions:
-
SCR1-4: Located at the N-terminus, responsible for the liquid phase regulatory function.
-
SCR19-20: Located at the C-terminus, specialized in recognizing host cell surface markers.
-
SCR6-8: Binds to C-reactive protein (CRP).
-
SCR5-7: Binds to apolipoprotein E (apoE).
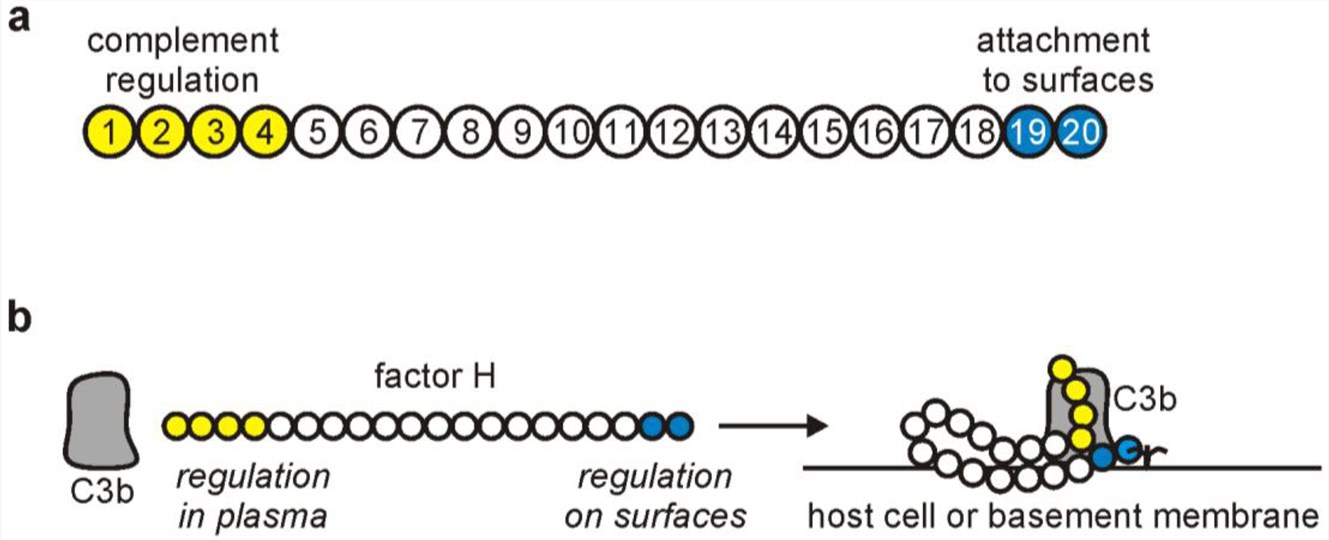 Fig.1 The schematic structure of factor H. 1,2
Fig.1 The schematic structure of factor H. 1,2
Functional Domains and Biological Roles
SCR1-4: Liquid phase regulatory core
-
C3b binding site: Inhibits the formation of the C3bBb enzyme complex by binding to C3b and blocking the binding of factor B to C3b.
-
Cofactor activity: Acts as a cofactor for factor I, promoting the degradation of C3b to iC3b and terminating the positive feedback loop.
-
Accelerated decay activity: Accelerates the irreversible decomposition of the formed C3bBb complex.
SCR19-20: Cell surface protection
-
Host marker recognition: Binds glycosaminoglycans (GAGs) and sialic acid to precisely localize host cell surface.
-
Pathological relevance: Anti-factor H antibodies often target SCR19-20, disrupting its binding ability and leading to uncontrolled complement bypass (e.g. aHUS).
Other functional domains
-
SCR6-8: Binds to CRP and regulates the inflammatory response.
-
SCR5-7: Binds to ApoE and may be involved in the regulation of lipid metabolism.
-
SCR18-20: Synergize with SCR1-4 to enhance C3b binding efficiency.
Function of Complement Factor H
The destructive potential of the complement system requires tight regulatory control to prevent host tissue damage while maintaining pathogen clearance. These mechanisms act at multiple levels, with CFH emerging as a central regulator of alternative pathways. The main functions of factor H can be categorized into several key roles.
Table 1 Functional roles of CFH in the complement system.
|
Function
|
Mechanisms
|
|
Cofactor Activity
|
CFH accelerates the decay of C3bBb, the alternative pathway C3 convertase, by acting as a cofactor for factor I. This results in the proteolytic inactivation of C3b.
|
|
Competing with Factor B
|
CFH competes with factor B for binding to C3b, thereby preventing the formation of the C3bBb convertase complex.
|
|
Protecting Host Cells
|
CFH binds to host cell surfaces, such as endothelial cells, epithelial cells, and some bacteria, helping to distinguish host from non-host surfaces. This prevents the complement system from attacking the body's own cells.
|
|
Regulation of the Alternative Pathway
|
As the key regulator of the alternative pathway, CFH maintains a delicate balance, ensuring that complement activation does not escalate into an inflammatory response that could be harmful to the body.
|
Factor H and C3b Receptor Interaction
One of the most critical interactions of CFH is with the C3b receptor. The C3b receptor (also known as CR1) is a membrane-bound protein that binds C3b fragments on the surface of pathogens and host cells. By binding to C3b, CFH helps prevent further complement activation on the host's cell surfaces, thus preventing immune-mediated damage. This interaction is vital in the regulation of the alternative pathway, ensuring that complement activity is limited to pathogen surfaces and does not extend to healthy host tissues.
-
Researchers have developed genetically modified mice, referred to as C3b receptor factor H mice, to investigate the regulatory role of CFH in the immune system. These mice express a mutant form of CFH that lacks the ability to bind to C3b receptors.
-
In C3b receptor factor H mice, the inability of CFH to bind C3b results in a hyperactive complement system, leading to excessive inflammation and tissue damage.
Complement Factor H Test
The CFH functional test evaluates the regulatory activity of CFH in the complement system, critical for diagnosing complement-mediated disorders like aHUS and C3 glomerulopathy (C3G).
The CFH functional test employs two primary approaches:
Enzyme-Linked Immunosorbent Assay (ELISA)
-
Sandwich ELISA: Purified human CFH is coated on plates, and patient serum is added to detect anti-CFH autoantibodies or quantify CFH levels.
-
Functional ELISA: Measures CFH's ability to act as a cofactor for Factor I-mediated cleavage of C3b to iC3b, using substrates like C3b-coated plates and factor I.
Hemolysis-Based Functional Assays
-
Reconstitution assays: Serum deficient in CFH is mixed with patient serum to assess restoration of hemolytic activity (e.g., CH50 or AH50). Purified CFH is added to confirm functional deficiency.
-
C3b cleavage assays: CFH's cofactor activity is tested by measuring factor I-dependent cleavage of C3b to iC3b, often using Western blot or ELISA.
Interpretation and Reference Ranges
-
Normal CFH activity: Sufficient to restore hemolytic activity in deficient serum or to effectively cleave C3b.
-
Insufficient CFH activity: <6% of normal indicates a genetic defect or severe dysfunction.
-
Elevated anti-CFH antibody: Suggests autoimmune aHUS.
Factor H and Disease Pathology
Abnormal function of CFH, a core regulatory protein of the complement system, is closely associated with a variety of diseases. By regulating the activation of the complement alternative pathway, CFH plays a key role in maintaining immune homeostasis and preventing host cell damage.
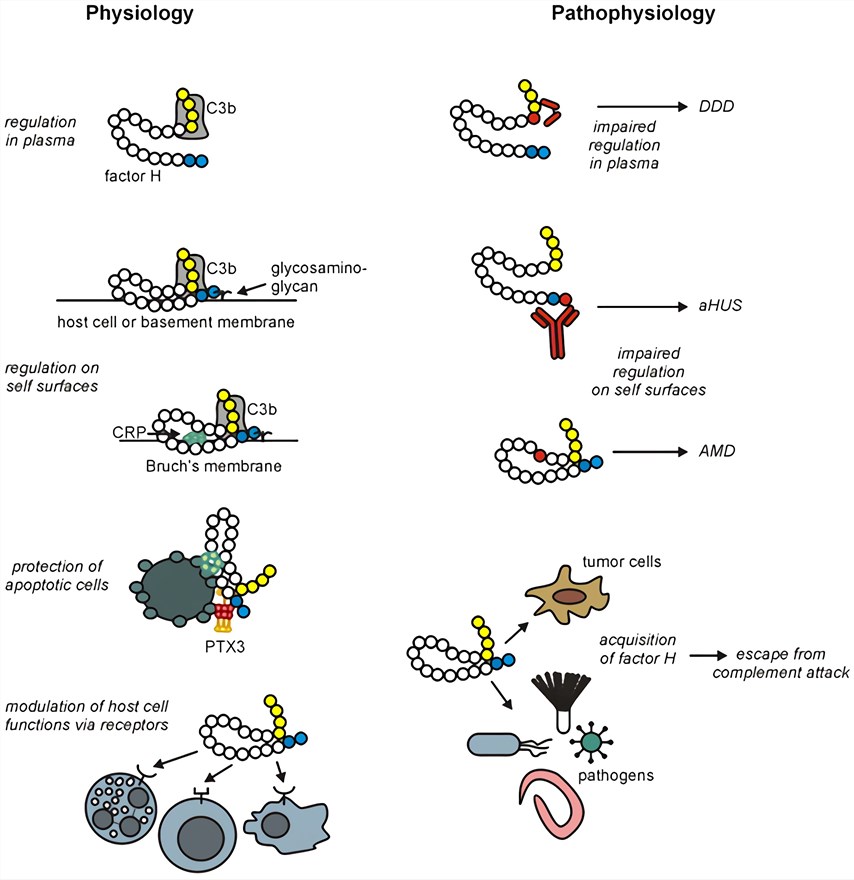 Fig.2 Overview of main factor H functions and their implication in pathological conditions. 1,2
Fig.2 Overview of main factor H functions and their implication in pathological conditions. 1,2
The role of CFH in disease is analyzed below in terms of different disease dimensions.
Table 2 Role of CFH in disease pathogenesis.
|
Disease Types
|
Specific Symptoms
|
Mechanism
|
|
Kidney diseases
|
aHUS
|
Anti-factor H antibodies primarily target the SCR19-20 region of CFH, disrupting its ability to bind C3b and leading to a loss of control of the complement alternative pathway.
|
|
C3G and membranous nephropathy
|
Decreased CFH concentration or activity (<6% of normal levels) leads to persistent activation of the complement alternative pathway and C3b deposition in the glomerular basement membrane, triggering inflammation and fibrosis.
|
|
Cardiovascular disease
|
Atherosclerosis and myocardial ischemia
|
Reduced or absent CFH causes the complement alternative pathway to go out of control, and the accumulation of C3b and C5b-9 triggers endothelial and cardiomyocyte injury.
|
|
Vascular wall inflammation and thrombosis
|
Abnormal CFH regulation leads to increased release of inflammatory mediators (e.g., C5a )
|
|
Autoimmune diseases
|
Autoimmune hepatitis (AIH)
|
Impaired CFH regulation leads to abnormal accumulation of C3b and C5b-9, exacerbating hepatocellular injury and chronic inflammation.
|
|
Systemic lupus erythematosus (SLE)
|
Decreased CFH levels are associated with SLE and may exacerbate immune complex deposition and organ damage through complement paracrine pathway activation.
|
|
Neurological disorders
|
Depression and schizophrenia
|
Abnormal activation of the complement system leads to neuronal damage, and deficits in CFH regulation may exacerbate synaptic clearance and neurodegenerative lesions.
|
|
Infectious diseases
|
Pathogens escape
|
Certain pathogens (e.g., meningococcus) evade complement killing by binding to the SCR19-20 region of CFH and camouflaging their own surface.
|
CFH plays a "protector" role in disease pathogenesis by regulating the complement alternative pathway, and its aberrant function directly leads to host cell damage and uncontrolled inflammation, making it a central target in a variety of diseases.
Factor H as a Therapeutic Target
Given its key role in immunomodulation, CFH offers an attractive target for the treatment of diseases associated with complement dysregulation. Several regulatory therapies are currently being explored, including strategies to enhance and inhibit CFH activity.
Enhancement of CFH activity in complement disease
-
CFH replacement therapy: In CFH-deficient mice, purified human CFH (hCFH) rapidly normalizes plasma C3 levels and resolves glomerular C3 deposits, mimicking dense deposit disease (DDD) and aHUS.
-
Immunosuppressive strategies: Reduce aFH antibody titers and supplement with CFH and factor I.
Inhibition of CFH in an immunosuppressive setting
-
Cancer immunotherapy: Tumors use CFH to evade complement-mediated lysis. Anti-CFH monoclonal antibody (mAb) disrupts this evasion, thereby enhancing tumor infiltration and C3d deposition by CD11b+ cells.
Table 3 Emerging treatment modalities.
|
Methods
|
Mechanisms
|
Clinical Significance
|
|
CFH-Ig fusion protein
|
Combines SCR1-7 (regulatory domain) with SCR19-20 (targeting) to extend half-life
|
Enhances CFH activity in renal disease
|
|
Small molecule inhibitors
|
Target CFH binding to C3b or host markers
|
Potential for oral treatment of chronic diseases
|
|
Gene therapy
|
Restoration of CFH expression in genetic defects
|
Addresses the underlying causes of inherited complement disorders
|
The dual role of CFH in immunomodulation and disease pathogenesis makes it a versatile therapeutic target. While CFH replacement and immunosuppression dominate the treatment of complement disorders, its inhibition in cancer and neurodegenerative diseases opens new avenues. Advances in CFH research hold great promise for the development of targeted therapies for the treatment of these diseases.
Creative Biolabs offers a full range of complement-related services and products, including:
If you want more information, please feel free to contact us.
References
-
Kopp, Anne, et al. "Factor h: a complement regulator in health and disease, and a mediator of cellular interactions." Biomolecules 2.1 (2012): 46-75. https://doi.org/10.3390/biom2010046
-
under Open Access license CC BY 3.0, without modification.
For Research Use Only.
Related Sections: