
Antitumor Activity of TfR-Targeted Peptide-Doxorubicin Conjugates
Introduction
The development of precision oncology therapeutics continues to confront a fundamental limitation: the narrow therapeutic indices of conventional chemotherapeutics. This challenge is exemplified by doxorubicin (DOX), a topoisomerase II inhibitor whose clinical utility is paradoxically constrained by its non-specific biodistribution. While DOX effectively disrupts DNA replication in rapidly dividing cells, its indiscriminate cytotoxicity extends to cardiomyocytes, hematopoietic precursors, and other healthy tissues, manifesting as dose-limiting cardiotoxicity, myelosuppression, and multi-organ dysfunction. These off-target effects not only reduce treatment tolerability but may also necessitate premature therapy termination, potentially undermining long-term remission.
Current strategies to circumvent this dilemma focus on leveraging tumor-specific molecular signatures. Among these, the transferrin receptor (TfR/CD71) emerges as a compelling ligand-receptor axis. As a key mediator of iron homeostasis, TfR is ubiquitously overexpressed in malignancies—a phenomenon driven by the elevated metabolic demands of proliferative cancer cells. This differential expression profile presents an exploitable vulnerability for selective drug delivery.
In this study, scientists designed and synthesized new peptide-doxorubicin conjugates (PDCs). They conjugated DOX to two different peptides, LT7 and its retro-inverso analog DT7 (hypothesized to offer improved TfR affinity and serum stability compared to LT7), via a disulfide bond linker. The resulting PDCs, LT7-SS-DOX and DT7-SS-DOX, aim to confine DOX activation to the reducing tumor microenvironment while preserving systemic stability. The results demonstrated that the PDCs exhibited targeted antiproliferative effects on TfR-overexpressing tumor cells, with DT7-SS-DOX showing higher stability and more potent in vitro activity.
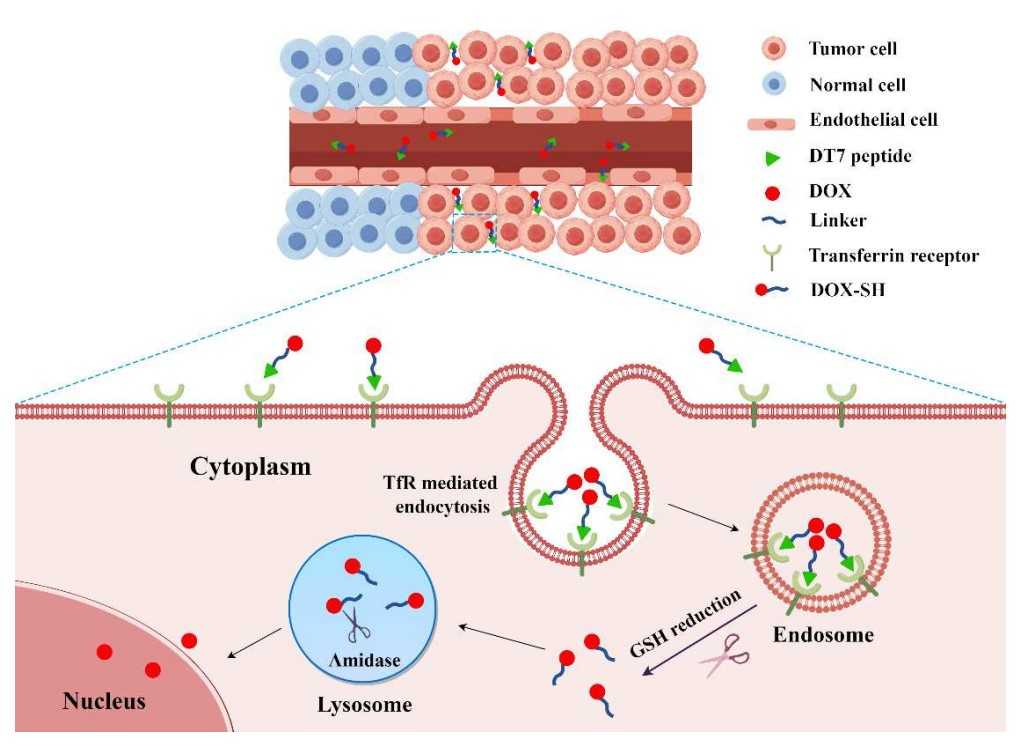 Fig.1 Schematic illustration of the mechanism of DT7-SS-DOX in TfR overexpressing tumor cells.1
Fig.1 Schematic illustration of the mechanism of DT7-SS-DOX in TfR overexpressing tumor cells.1
Serum Stability and Drug Release of PDCs
To evaluate the potential of the PDCs for in vivo applications, their serum stability and drug release behavior were examined. DT7-SS-DOX demonstrated higher serum stability and a sustained drug release profile compared to LT7-SS-DOX, indicating that the retro-inverso peptide modification enhanced the stability of the conjugate.
- Serum Stability assay: The conjugates were incubated in mouse serum, and their stability over time was assessed by measuring the amount of intact conjugate remaining. LT7-SS-DOX exhibited rapid degradation kinetics, with near-complete degradation (≥95%) by 1 h. In contrast, DT7-SS-DOX demonstrated markedly enhanced serum persistence, with detectable intact conjugate (∼15%) remaining at 24 h.
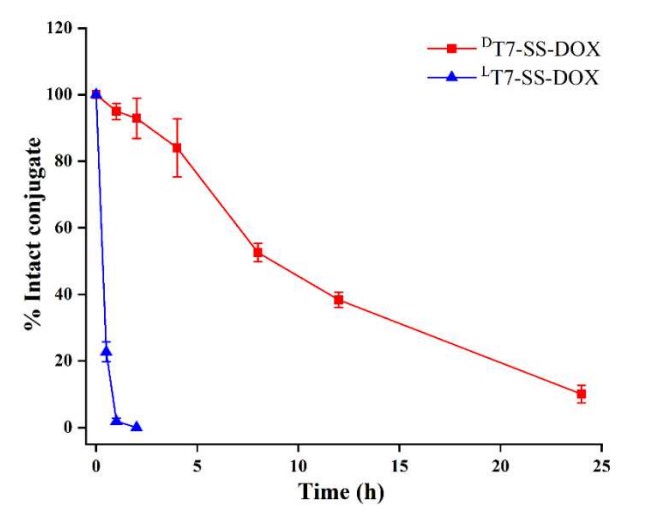 Fig.2 Stability analysis of PDCs in mouse serum.1
Fig.2 Stability analysis of PDCs in mouse serum.1
- Drug Release assay: The redox-responsive drug release behavior of the PDCs was investigated by co-incubating them with varying concentrations of glutathione (GSH). Results indicated that DT7-SS-DOX exhibited slower degradation than LT7-SS-DOX in both 5 mM and 5 μM GSH. This stimulus-responsive behavior suggests that the slower release profile of DT7-SS-DOX in the tumor microenvironment could promote extended intratumoral drug retention, while its stability under physiological conditions may minimize off-target toxicity. This approach addresses limitations associated with traditional disulfide linkers and potentially broadens the therapeutic window.
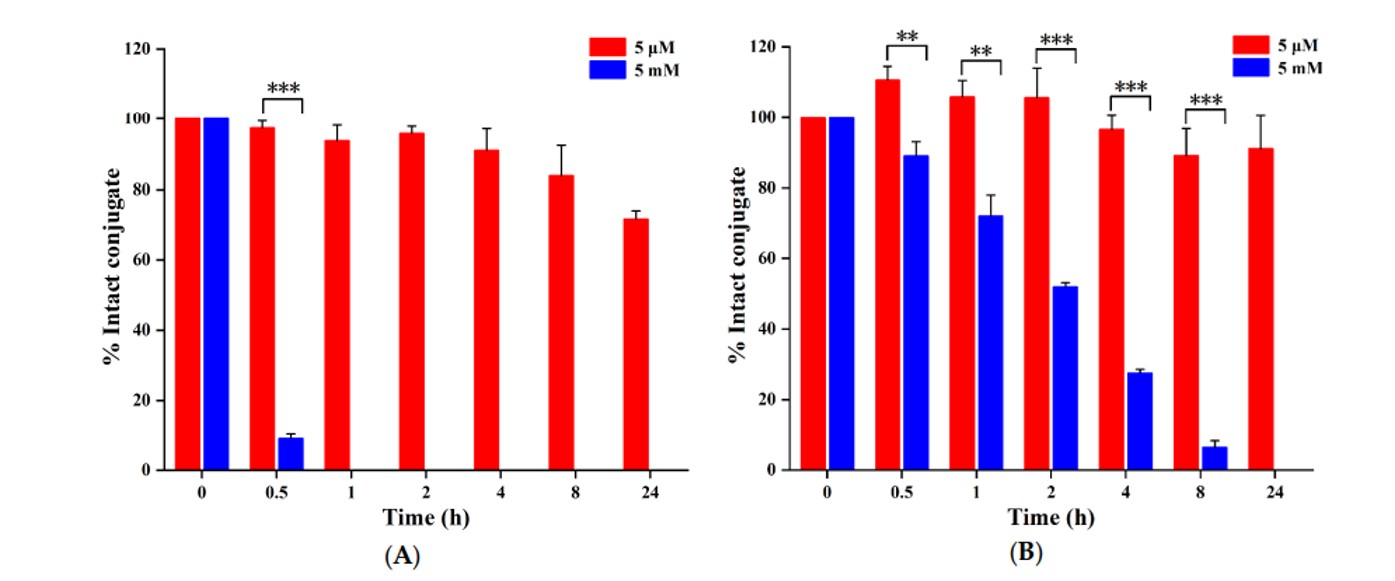 Fig.3 Reduction-triggered degradation of (A) LT7-SS-DOX and (B) DT7-SS-DOX by GSH.1
Fig.3 Reduction-triggered degradation of (A) LT7-SS-DOX and (B) DT7-SS-DOX by GSH.1
Cellular Uptake Analysis of PDCs
The cellular uptake of the PDCs was investigated using confocal microscopy. Tumor cells overexpressing TfR were incubated with fluorescently labeled PDCs, and the intracellular localization of the conjugates was visualized. The result revealed TfR-dependent uptake of PDCs (DT7-SS-DOX/LT7-SS-DOX) in tumor cells, with minimal accumulation in normal cells, contrasting with the non-specific uptake of free DOX. Competitive inhibition assays confirmed TfR-mediated endocytosis as the primary uptake pathway. The DT7-SS-DOX conjugate exhibited enhanced tumor cell uptake compared to LT7-SS-DOX, attributed to its superior TfR-binding affinity.
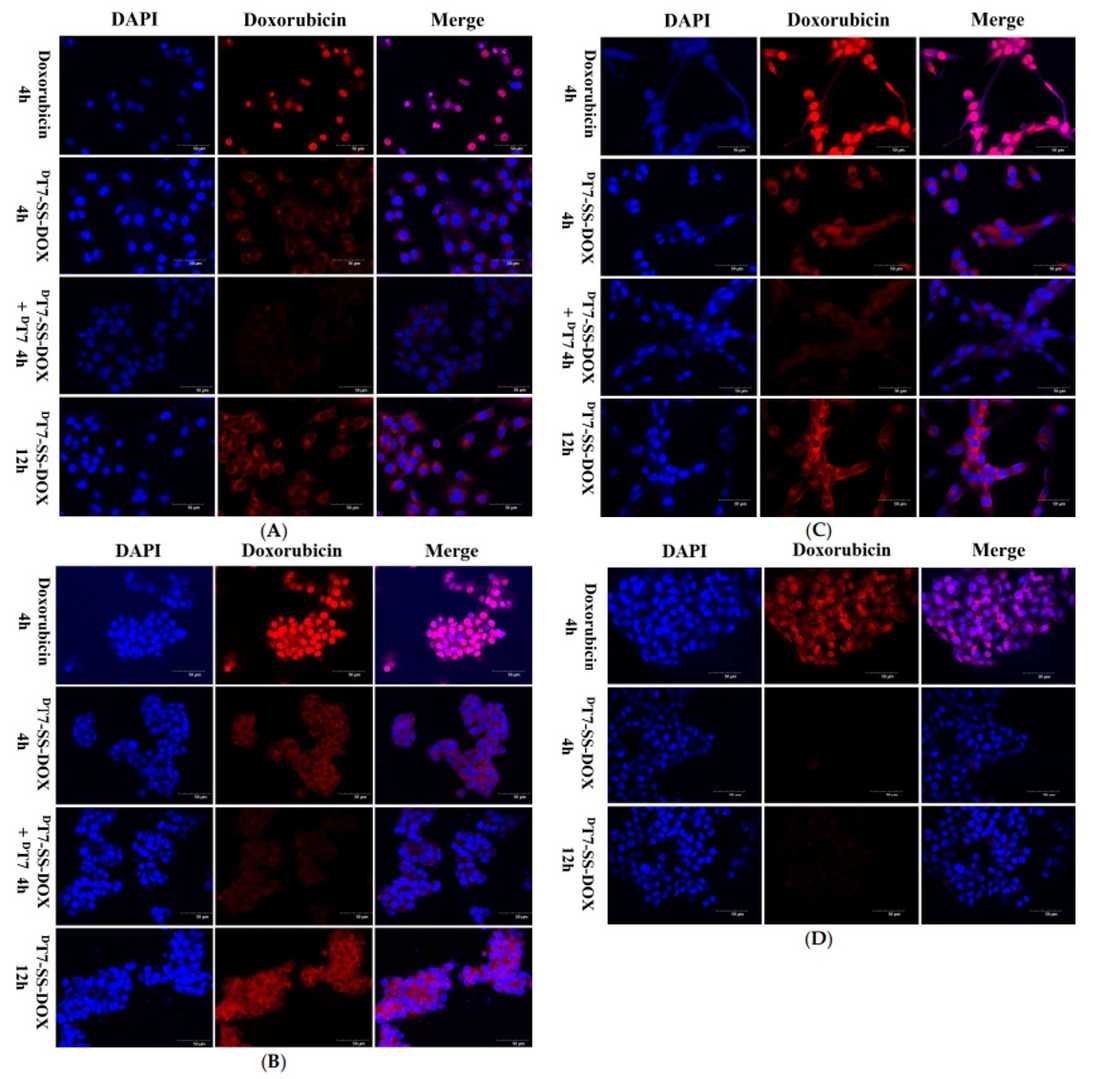 Fig.4 Free DOX (10 µM, 4 h) and DT7-SS-DOX (10 µM DOX eq., 4 & 12 h) visualized in (A) A549, (B) HepG2, (C) U87, and (D) LO2 cells via confocal microscopy.1
Fig.4 Free DOX (10 µM, 4 h) and DT7-SS-DOX (10 µM DOX eq., 4 & 12 h) visualized in (A) A549, (B) HepG2, (C) U87, and (D) LO2 cells via confocal microscopy.1
In Vitro Cytotoxicity of PDCs
CCK-8 assays evaluated the cytotoxicity of free DOX, LT7-SS-DOX, and DT7-SS-DOX against TfR-overexpressing tumor cells and TfR-low normal cells. Free DOX exhibited potent, non-selective cytotoxicity, while PDCs demonstrated tumor-selective activity with significantly reduced toxicity in normal cells. LT7-SS-DOX displayed weaker antiproliferative effects, attributed to lower TfR affinity and rapid degradation. Notably, delayed cytotoxicity was observed with both conjugates, which is linked to TfR-mediated endocytosis and subsequent intracellular release of DOX via disulfide cleavage and amidase activity. These findings underscore the potential of TfR-targeted PDCs to decouple antitumor efficacy from systemic toxicity.
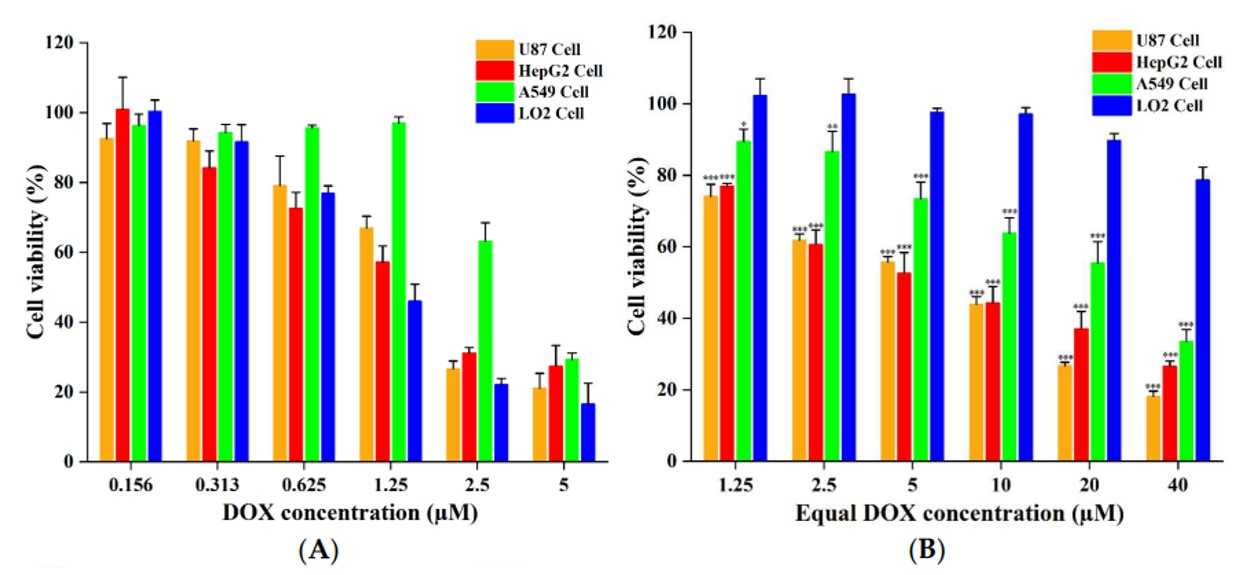 Fig.5 In vitro cytotoxicity of (A) free DOX and (B) DT7-SS-DOX against cells.1
Fig.5 In vitro cytotoxicity of (A) free DOX and (B) DT7-SS-DOX against cells.1
In conclusion, this study successfully designed and synthesized TfR-targeted PDCs with enhanced selectivity and efficacy against tumor cells. The use of retro-inverso peptides improved the stability and drug release characteristics of the conjugates. These findings suggest that this targeted drug delivery strategy holds promise for improving cancer treatment outcomes.
With our specialized knowledge in bioconjugation, Creative Biolabs offers peptide-small molecule conjugation services, enabling researchers to create novel targeted therapeutics for various applications. Contact us for more details.
Reference
- Yu, Jiale, et al. "New Transferrin Receptor-Targeted Peptide–Doxorubicin Conjugates: Synthesis and In Vitro Antitumor Activity." Molecules 29.8 (2024): 1758. Under open access license CC BY 4.0, without modification.