Creative Biolabs is offering the most comprehensive services for antibody development projects. With strict regulation and effective execution, we are dedicated to providing the most valuable solutions to complete your projects.
HighlightsWith over a decade of experience in phage display technology, Creative Biolabs can provide a series of antibody or peptide libraries that are available for licensing or direct screening. These ready-to-use libraries are invaluable resources for isolating target-specific binders for various research, diagnostic or therapeutic applications. Highlights
Creative Biolabs has established a broad range of platforms for developing novel antibodies or equivalents. These cutting-edge technologies enable our scientists to meet your demands from different aspects and tailor the most appropriate solution that contributes to the success of your projects.
HighlightsWith deep understanding in antibody-related realms and extensive project experience, Creative Biolabs offers a variety of references to help you learn more about our capacities and achievements, including infographic, flyer, case study, peer-reviewed publications, and all kinds of knowledge that can assist your projects. You are also welcome to contact us directly for more specific solutions.
HighlightsGet a real taste of Creative Biolabs, one of the most professional custom service providers in the world. We are committed to providing highly customized comprehensive solutions with the best quality to advance your projects.
HighlightsDedicated to bringing differentiated and high-quality services to our global clients, Creative Biolabs is a highly proved provider of protein engineering services. With the values of customer focus and respect, we employ the well-trained personnel to provide our customers with the most innovative technologies, valuable services and tailor-made solutions.
In recent years, computer-aided or in silico methods have played an increasingly important role in the protein or enzymatic engineering. These computational methods range from bioinformatics analysis of primary sequences to computer simulation of tertiary structures to prediction of new structures by de novo design, which provides new strategies for improving enzyme activity, specificity and stability. Creative Biolabs has extensive experience in bioinformatics analysis, computer simulation and de novo design. By establishing a series of advanced platforms, we provide these computer-aided analyses to speed up the process, reduce surprises and predict the properties, thereby reducing the cost of R&D.
Bioinformatics tools can be used to identify interaction hotspots and restrict the alphabet of substitutions for the design of smart libraries in focused directed evolution. Feng et al. developed the Adaptive Substituent Reordering Algorithm (ASRA), which can be used in combination with many directed evolution methods. ASRA can identify the underlying regularity of the protein property and predict the properties of uncharacterized proteins (Fig.1). Moreover, ASRA does not require assumptions of linearity, additivity, or any functional form of structure-property relationships. Thus, this method provides a reliable prediction of multiple mutants with improved enantioselectivity. The method can also be applied to other properties such as enzyme activity and stability.
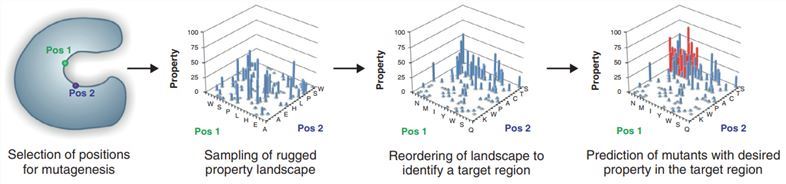 Fig.1 Workflow for adaptive substituent reordering algorithm identifying mutants with desired properties. (Damborsky, 2014)
Fig.1 Workflow for adaptive substituent reordering algorithm identifying mutants with desired properties. (Damborsky, 2014)
The empirical valence bond modeling technique can be used for the quantitative analysis of the enantioselectivity of some enzymes, such as lipases. For example, the researchers evaluated the kcat and kcat/Km of the individual enantiomers of 4-nitrophenyl 2-methylheptanoate by this method. In addition, other methods, such as molecular docking, molecular dynamics, and quantum mechanical calculations, are also commonly used to rationally design enzymes to evaluate the enantioselectivity of the enzyme.
The design of biocatalysts that bind small ligands with good affinity requires the precise calculation of rather weak protein-ligand interactions. Therefore, an optimized binding site must offer: 1) specific favorable hydrogen-bonding and van der Waals interactions with the ligand; 2) high overall shape complementarity to the ligand; 3) structural pre-organization in the unbound protein state to minimize entropy loss upon ligand binding. Fig.2 shows the first successfully de novo design of ligand-binding protein with a low-to-mid micromolar range. Moreover, de novo design can be combined with techniques such as yeast surface display and fluorescence-activated cell sorting to further optimize the design to obtain proteins with high binding affinity and high selectivity.
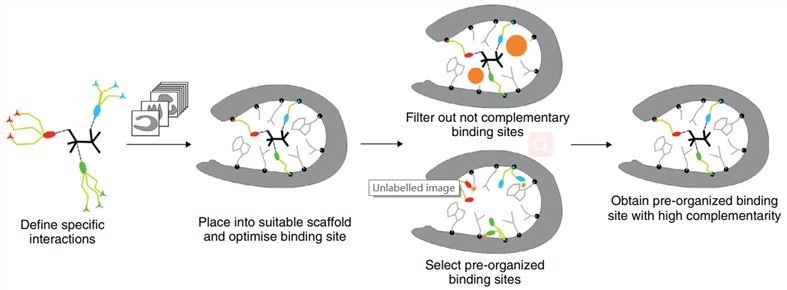 Fig.2 Workflow for de novo design of ligand-binding protein. (Damborsky, 2014)
Fig.2 Workflow for de novo design of ligand-binding protein. (Damborsky, 2014)
Through our advanced technology and professional knowledge, we can provide best-in-class computer-aided enzyme selectivity analytical services to clients around the world. As a global total quality assurance provider within the industry, we are characterized by:
Any of our service packages can be booked on its own, or in conjunction with others. Please contact us for project quotations and more detailed information.
Reference
All listed services and products are For Research Use Only. Do Not use in any diagnostic or therapeutic applications.