Creative Biolabs has a tradition of commitment. To achieve efficient execution and regulatory approval, we offer careful considerations of your program for the development of a cellular or gene therapy product – now and in the future.
EXPLORE MORE HighlightsWe focus on unmet needs and develop novel cellular and gene drugs and solutions that offer significant benefits over existing options.
EXPLORE MORE HighlightsThe advent of Chimeric Antigen Receptor T-cell (CAR-T) therapies has revolutionized the field of oncology, providing new avenues for treating various forms of cancer. Our 20 years of experience in the biotechnology sector have equipped us with the expertise and technological capabilities to support the entire lifecycle of CAR-T products, from early development to commercialization.
EXPLORE MORETo accelerate advanced breakthroughs of your projects, we offer broad range of platforms which enable our clients be free to tackle problems with cutting-edge technologies from different angles and in different methods.
EXPLORE MORE HighlightsUse the resources in our library to help you understand your options and make critical decisions for your study. We offer oncolytic virus, CAR-T, and dendritic cell related documents, as well as newsletter. If you don't find the answers you're looking for, contact us for additional assistance.
EXPLORE MORE HighlightsGet a real taste and understanding of the business and culture of one of the world's great research-based cellular and gene therapy discovery and development companies.
EXPLORE MORE
Immunotherapy, which consists of gene-edited immune cells, plays an important role in cancer treatment. This "live" therapy, which targets tumor antigens, has been successful in a growing number of clinical studies in the form of a lasting response in many cancer patients. In adoptive cell transfer (ACT), immune cells, especially T cells, involved in the anti-tumor response are collected from a cancer patient, amplified in vitro and selected or modified, and then given back to the patient.
Dendritic cells are the most specialized antigen-presenting cells(APCs) for triggering innate and adaptive immune responses. These cells can activate CD4+ T cells through MHC-II antigen presentation and CD8+ T cells through a process called "cross-presentation" (presentation of exogenously captured antigens on MHC-I). Gene-edited dendritic cells (DCs) are also widely used in combination therapy, especially in combination with checkpoint inhibitors (CIs), to break immune tolerance and enhance immune response to the tumor immunosuppressive environment.
In lung cancer therapy, NK cells can bridge and coordinate innate and "downstream" adaptive immune responses, making them an ideal platform for new cancer therapies. However, it’s difficult to activate NK cells, lack of tumor-specific NK cells, up-regulated checkpoint pathway expression, and low mutation burden hinder the development of long-term adaptive immunity. Therefore, combining NK cell reactivation strategies with other therapies may benefit clinical outcomes in cancer patients.
Genetically engineered DC can highly stimulate the accompanying antigen-specific CD8+ and CD4+ T cell responses without affecting the MHC-I epitope presentation pathway. Genetically modified dendritic cells are usually administered with anti-CTLA-4 monoclonal antibody (mAb). In a phase II study, dendritic cells electroperforated with synthetic mRNA (TriMixDC-MEL). The TriMix-DC mixture was co-electroporated with a melanoma-associated antigen associated with HLA Class II targeting signals (gp100, tyrosinase, MAGE-A3, or MAGE-C2 fusion to DC). Ipilimumab, a CTLA-4 blocking monoclonal antibody, produced a tolerable and highly effective antitumor response. In another clinical study, gp100 and tyrosinase-loaded DC induced clinical responses in patients with stage III and IV melanoma when administered in combination with Ipilimumab. In a phase I study, dose-increasing systemic therapy with the anti-CTLA-4 mAb Tremelimumab in combination with biweekly vaccination with MART-1-containing DC achieved a 25% response rate, which was significantly higher than Tremelimumab monotherapy (~10%) and DC vaccination monotherapy (~15%).
Researchers sorted bone marrow CD34+ progenitors and T cells, and transduced them with anti-CD33 41BBz CAR lentivector. Transduced T (CAR T) and CD34+ progenitors were also sorted three days after transduction. By using phenotypic analysis flow cytometry, it was found that CD141/CLEC9A in the CAR-DC is significantly higher than control DC, which suggests that the activation of 4-1BB induces CD34+ progenitor cells to intra tumor DC. These cytotoxicity tests showed a death rate of 63.2% for Kasumi-1 caused by CAR T/CAR DC. Only CAR T cells were 3.5%. CAR T/CAR-DC also showed more annexin V positive Kasumi-1 compared with the CAR-T and control T cells. These cytotoxicity tests showed that CAR DC enhanced the anti-Kasumi-1 cytotoxicity of anti-CD33 CAR-T cells.
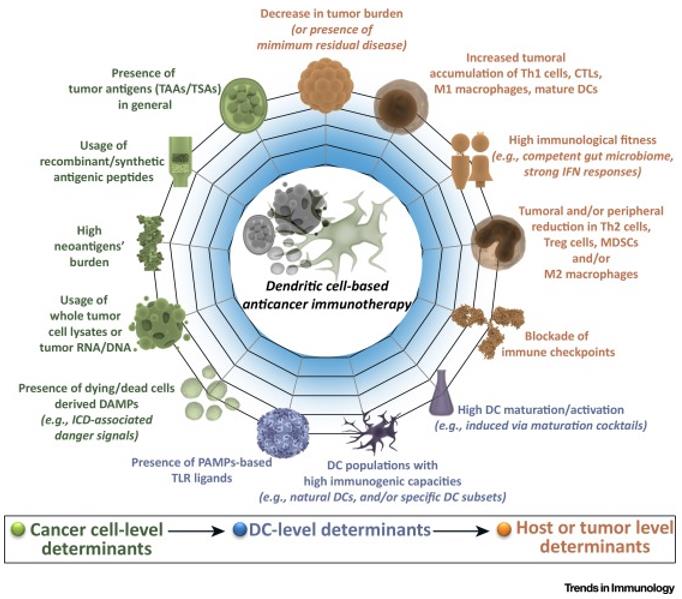
Autologous CD14+ mononuclear cells from peripheral blood of cancer patients (by leukocyte separation) were differentiated into immature DCs using GM-CSF and IL-4/IL-13 in vitro. The immature DC is then provided with tumor antigens that can be derived from a variety of sources, namely intact autologous/allogenic cell lysates (inducing cell death; further antigen processing is required), recombinant antigenic peptides (from tumor-associated antigens or neoantigens; no antigenic processing is required), RNA/DNA from cancer cells, recombinant RNA/DNA encoding specific tumor antigens, and DCs/ tumor fusion hybrids (which provide stable antigen presentation). These antigen-loaded DCS are further matured/stimulated by a "maturation mixture" composed of specific cytokines, primarily TNF/IL1b/IL6/PGE2; Although PGE2 represents a double-edged sword, as it not only induces DC lymph node homing, but also promotes immunosuppressive Treg cells through IL12p70 inhibition/ indoleamine 2,3 dioxygenase (IDO). PGE2-related problems can be overcome with toll-like receptor (TLR) agonists, although they can induce IDO. These factors promote DC-based MHC I/MHC II driven antigen cross-presentation, the presence of appropriate costimulation (CD80/CD86/CD83/CD40), and upregulation of lymph node homing (via CCR7). These fully mature immunogenic DCS are then transfused back into the patient, where they migrate to the nearest lymph node and present cancer antigens to CD4+/CD8+ T cells in the presence of co-stimulating and T-cell stimulating cytokines that promote anti-tumor T cell polarization. For example, Type 1/17 polarization (Th1/Th17) and cytotoxic T lymphocyte (CTL) activity.
In existing phase I/II clinical trials, adenovirus-transfected dendritic cells expressing MUC1 and survival proteins were studied in combination with cytokine-induced killer cells (CIKs). Clinical efficacy before and after treatment (objective response rate: 39%, disease control rate: 75%) and safety evaluation of DC-CIK confirm the feasibility of this combination therapy in patients with advanced renal cancer. Successful cases of combining modified DCS with chemotherapy and radiotherapy have been reported, demonstrating the powerful synergistic effect of engineered dendritic cells on combination therapy.
In a Phase I clinical study to evaluate the safety and immune response induced by direct injection of autologous immature DCs into the tumor during radiotherapy in patients with advanced liver cancer, patients with advanced/metastatic liver cancer who were not candidates for surgery or transarterial embolization were enrolled. The patient group received two vaccinations. Each inoculation consisted of intratumoral injection of autologous immature DC into four dose groups 2 days after a single conformal radiotherapy of 8 Gy. The second dose was given 3 weeks later. Of the 14 patients admitted, 12 completed two cycles of vaccination. The treatment was well tolerated at any dose level. Six patients developed mild transient fever (grade 1-2) with chills, three patients developed grade 1 fatigue, and one patient developed mild myalgia and arthralgia after DC injection. There is no clinical evidence of autoimmune disease. There were two partial responses and four minor responses. Alpha-fetoprotein (AFP) levels fell by more than 50 percent in three patients. Assessment of immune response was completed in 10 patients at 2 weeks after the second vaccination cycle. AFP specific immune responses were detected in 8 patients by cytokine release assay and in 7 patients by ELISPOT assay. Six patients showed increased cytotoxic activity of NK cells after vaccination. These data suggest that the combination of intratumor injection of dendritic cells and conformal radiotherapy is safe and can induce tumor-specific and innate immunity.
A total of 82 patients with stage III and 137 patients with stage IV melanoma received dendritic cell vaccine monotherapy between June 1999 and July 2015. This is done primarily through intranasal administration (64%), and the most common antigen loading method is mRNA electroporation (52%, Table 1). All stage III patients received moDC, while 79% of patients with stage IV disease received moDC, 11% received pDC, and 10% received mDC. A total of 59% of stage III patients and 10% of stage IV patients completed all 3 cycles of vaccination, 20% of stage III patients and 7% of stage IV patients received 2 cycles, and 22% of stage III patients and 83% of stage IV patients received only 1 cycle. Median follow-up time from apheresis to death or examination was 54.3 months for stage III patients (range 3.7 -- 162.4 months) and 12.9 months for stage IV patients (2.0 -- 179.7 months).
References
For any technical issues or product/service related questions, please leave your information below. Our team will contact you soon.
All products and services are For Research Use Only and CANNOT be used in the treatment or diagnosis of disease.
 NEWSLETTER
NEWSLETTER
The latest newsletter to introduce the latest breaking information, our site updates, field and other scientific news, important events, and insights from industry leaders
LEARN MORE NEWSLETTER NEW SOLUTION
NEW SOLUTION
CellRapeutics™ In Vivo Cell Engineering: One-stop in vivo T/B/NK cell and macrophage engineering services covering vectors construction to function verification.
LEARN MORE SOLUTION NOVEL TECHNOLOGY
NOVEL TECHNOLOGY
Silence™ CAR-T Cell: A novel platform to enhance CAR-T cell immunotherapy by combining RNAi technology to suppress genes that may impede CAR functionality.
LEARN MORE NOVEL TECHNOLOGY NEW SOLUTION
NEW SOLUTION
Canine CAR-T Therapy Development: From early target discovery, CAR design and construction, cell culture, and transfection, to in vitro and in vivo function validation.
LEARN MORE SOLUTION