Design of siRNA Therapeutics
Introduction
Designing a therapeutic small interfering RNA (siRNA) is not simply a matter of finding a 21-nucleotide sequence that matches a messenger RNA. A development-ready candidate must combine efficient RNA-induced silencing complex loading, durable target knockdown, low seed-mediated off-target activity, acceptable innate immune stimulation, manufacturable chemistry, and a delivery format suited to the target tissue. A decision-oriented framework is provided for translating a biological target into a testable siRNA lead, with particular attention to the interplay among sequence selection, chemical modification, and delivery strategy that distinguishes therapeutic development from routine research knockdown. The discussion also outlines how staged in vitro screening can narrow the candidate pool before animal evaluation.
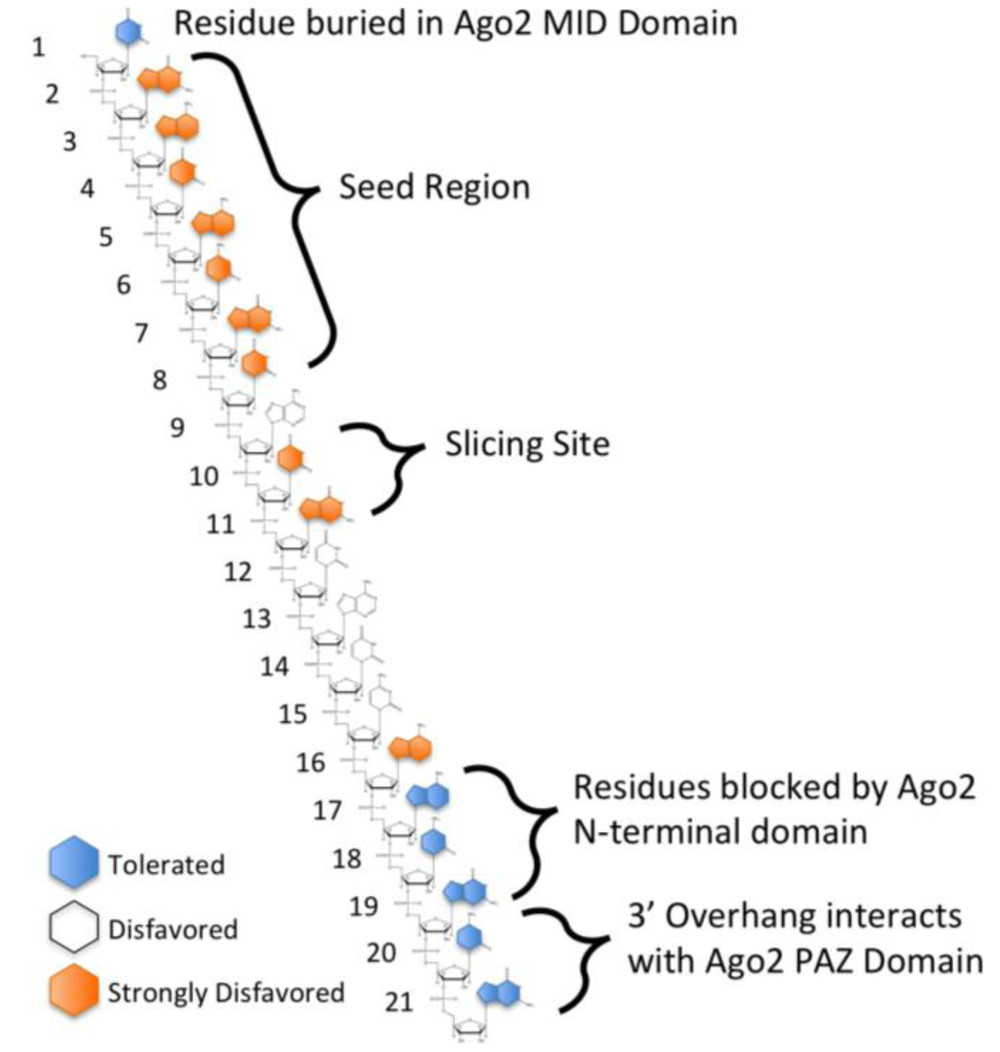 Figure 1. Tolerance for mismatches between an siRNA and its target.1
Figure 1. Tolerance for mismatches between an siRNA and its target.1
Start with a Therapeutic Product Profile
The first design decision should be the intended product, not the oligonucleotide sequence. Tissue, route, dosing interval, target-cell abundance, disease kinetics, and acceptable safety margins determine which siRNA architecture is realistic. A sequence optimized without these constraints may perform well in transfection assays yet fail when conjugated, formulated, or evaluated at clinically relevant exposure.
Define the biological intervention
- Confirm that reducing the target transcript is expected to improve disease biology and determine whether partial, near-complete, transient, or sustained knockdown is required.
- Specify the disease-relevant transcript, isoform, splice junction, mutation, or allele. A target shared by beneficial and pathogenic isoforms may require a different strategy from an allele-selective program.
- Identify the pharmacologically accessible cell population. Hepatocytes are compatible with receptor-mediated conjugates, whereas tumors, immune cells, muscle, eye, and central nervous system tissues present different uptake and endosomal barriers.
- Define a measurable pharmacodynamic chain: siRNA exposure, target mRNA reduction, target protein reduction, pathway modulation, and disease-relevant functional effect.
Translate the profile into design constraints
| Product question | Design consequence | Early evidence needed |
|---|---|---|
| Where must the siRNA act? | Select a delivery class and evaluate target-cell receptor expression, extracellular stability, tissue penetration, and intracellular trafficking. | Cell uptake, subcellular localization, biodistribution, and target engagement |
| How long should silencing last? | Balance nuclease-resistant chemistry and tissue retention against reversibility and accumulation. | Time-course of mRNA/protein recovery and repeat-dose response |
| How much knockdown is sufficient? | Set a potency threshold linked to pathway rescue rather than maximizing knockdown without context. | Concentration–response and functional biomarker relationship |
| How selective must the candidate be? | Apply transcriptome, seed-match, paralog, isoform, and species-homology filters before synthesis. | RNA profiling, orthogonal confirmation, and mismatch controls |
Build a Sequence Selection Funnel for siRNA Therapeutics
A practical program begins with broad computational enumeration and progressively applies biological, biophysical, and safety filters. No single design rule reliably predicts therapeutic performance because sequence, chemical modification, target-site accessibility, Ago2 loading, and assay context interact. The objective is therefore to generate a diverse but controlled panel that can be ranked experimentally rather than selecting one apparently optimal duplex.
1. Define the targetable transcript space
- Use the current reference transcript and confirm clinically relevant isoforms, exon structure, untranslated regions, coding sequence, single-nucleotide variants, RNA editing sites, and common population polymorphisms.
- Exclude regions with uncertain annotation, repeated sequence, extensive homology to unintended transcripts, or variants likely to disrupt binding in the intended population.
- For allele-selective designs, place the discriminating nucleotide at positions where mismatch sensitivity can be tuned, then test both mutant and wild-type transcripts under matched conditions.
2. Rank duplex candidates by complementary criteria
| Design dimension | Favorable direction | Reason for inclusion |
|---|---|---|
| Guide-strand loading | Thermodynamic and structural asymmetry that favors intended guide incorporation | Reduces passenger-strand loading and improves productive RISC formation |
| Target accessibility | Avoid strongly structured or protein-occluded transcript regions when evidence is available | Hybridization must compete with native RNA folding and RNA-binding proteins |
| Sequence composition | Moderate GC content; avoid long homopolymers, internal repeats, and synthesis-prone motifs | Supports duplex behavior, synthesis quality, and candidate diversity |
| Off-target liability | Minimize perfect near-matches and high-risk seed complementarity in expressed transcripts | Ago-mediated cleavage and miRNA-like seed repression can produce different off-target phenotypes |
| Immune-recognition motifs | Flag sequence contexts associated with endosomal or cytosolic RNA sensing | Sequence and chemistry jointly influence cytokine and innate immune responses |
3. Preserve panel diversity
The initial synthesis set should include candidates distributed across multiple transcript regions and sequence-feature clusters. Closely related candidates often fail for the same hidden reason, so a panel of twenty diverse duplexes can be more informative than twenty variants of one favored site. For therapeutic projects, custom siRNA synthesis should maintain traceable strand identity, purity, duplexing conditions, mass confirmation, and a chemistry map that can be carried forward into screening.
Engineer the Chemical Architecture for siRNA Therapeutics
Chemical modification transforms an easily degraded RNA duplex into a drug-like molecule, but modifications are positional design elements rather than interchangeable decorations. A pattern that stabilizes one sequence may alter Ago2 compatibility, strand selection, target cleavage, or off-target behavior in another. Consequently, sequence and chemistry should be co-optimized in a small matrix rather than treated as independent steps.
Core medicinal-chemistry functions
- Ribose modifications such as 2'-O-methyl and 2'-fluoro can improve metabolic stability and reduce recognition by innate immune sensors, while their positions influence RISC activity.
- Terminal phosphorothioate linkages can protect vulnerable ends and influence protein binding, distribution, and tolerability; excessive use may create liabilities rather than additional benefit.
- Guide 5'-end configuration must remain compatible with Ago loading. Stabilized phosphate mimics can support activity when native phosphorylation is inefficient or unstable.
- Passenger-strand modifications can reduce passenger loading, while selected guide seed modifications may attenuate miRNA-like off-target repression without eliminating on-target cleavage.
- Conjugation handles, linkers, and end-group orientation must be incorporated early because they can change duplex stability, serum binding, cellular uptake, and endosomal release.
Use a chemistry matrix, not a single "fully modified" design
| Variant | Purpose | Key comparison |
|---|---|---|
| Baseline therapeutic pattern | Establish a stable, RISC-compatible starting point | Potency, serum stability, cytokine response |
| Seed-tuned guide | Reduce seed-mediated transcript repression | On-target IC50 versus transcriptome off-target burden |
| Passenger-suppressed duplex | Bias strand selection and reduce passenger activity | Guide/passenger reporter assays and Ago loading |
| Conjugation-ready duplex | Test the exact end, linker, and chemistry needed for delivery | Free duplex versus conjugated uptake and potency |
| Stability-adjusted duplex | Extend or shorten exposure for the intended dosing interval | Metabolic profile, tissue half-life, duration of knockdown |
Match the Molecule to the Delivery Route
Delivery should be considered during sequence and chemistry selection because the same siRNA can behave differently as a free duplex, ligand conjugate, lipid nanoparticle cargo, polymer complex, or vector-expressed hairpin. The useful question is not which delivery technology is best in general, but which system produces sufficient cytosolic guide-strand exposure in the relevant cells at a tolerable dose.
Delivery archetypes
- Receptor-targeted conjugates are attractive when the target cell expresses an abundant, recycling receptor. GalNAc conjugation is established for hepatocyte delivery through the asialoglycoprotein receptor, whereas extrahepatic receptors require case-specific validation.
- Lipid and other nanoparticles can protect siRNA, carry higher payloads, and enable local or systemic administration, but organ distribution, innate immune activation, particle heterogeneity, and endosomal escape require optimization.
- Antibody-, peptide-, aptamer-, and oligonucleotide-based conjugates may add cell selectivity, yet conjugate orientation, linker stability, receptor trafficking, and release mechanisms must be evaluated together.
- Local administration can reduce systemic exposure for ocular, pulmonary, dermal, or intratumoral applications, although tissue diffusion and residence time may become the dominant limitations.
A delivery-development plan should compare uptake with productive silencing rather than relying on total cell-associated fluorescence. Functional delivery requires escape from endosomes and access to Ago2 in the cytosol. For programs that have not yet selected a carrier, RNAi delivery method development can be structured around the target cell, administration route, dose ceiling, and desired duration of action.
Screen for Potency, Selectivity, and Developability
Therapeutic lead selection requires a cascade that separates sequence activity from transfection artifacts and tests whether knockdown is mechanistically linked to the desired phenotype. The most informative workflow narrows candidates in stages, introduces the intended chemistry and delivery format early, and uses orthogonal assays before animal testing.
Tier 1: sequence and concentration response
- Test multiple concentrations and include non-targeting, mock, positive-control, and delivery-only controls.
- Measure target mRNA at an appropriate time point and confirm protein reduction after accounting for protein half-life.
- Compare at least two independent active siRNAs against the same target; shared phenotype strengthens target-level interpretation.
- Use a rescue construct or mismatch control when feasible to distinguish target-dependent effects from sequence-specific toxicity.
Tier 2: delivery-relevant and safety-focused assays
- Retest shortlisted sequences using the intended modification pattern and formulation or conjugate rather than extrapolating from unmodified transfection data.
- Assess viability, cell stress, cytokines, complement-related signals where relevant, hemocompatibility, and tissue-specific toxicities at multiples of the effective concentration.
- Characterize serum and tissue-matrix stability, metabolite patterns, protein binding, uptake, intracellular localization, and duration of knockdown.
- Use RNA profiling to distinguish intended pathway changes from broad seed-mediated or stress-response signatures.
Transcriptome-level analysis is most useful when interpreted alongside predicted seed matches, target expression, concentration dependence, and phenotypic rescue. A broad expression shift at doses far above the pharmacologic range should not be weighted the same as a reproducible off-target program near the expected exposure. Purpose-designed RNA profiling can therefore support both candidate ranking and mechanism-of-action interpretation.
Advance Candidates with Translation in Mind
An siRNA lead should enter in vivo studies only after its sequence identity, chemical pattern, formulation, analytical quality, potency window, and principal in vitro liabilities are defined. Species selection deserves particular attention because target-site conservation, receptor expression, immune sensing, and oligonucleotide disposition may differ substantially between human cells and animal models.
Lead nomination checklist
| Decision area | Minimum lead-level evidence | Common reason to pause |
|---|---|---|
| Target engagement | Reproducible mRNA and protein reduction with a concentration–response relationship | Knockdown measured only at one high concentration |
| Mechanistic function | Disease-relevant phenotype linked to target suppression and supported by controls | Phenotype without orthogonal or rescue evidence |
| Specificity | Sequence-based risk assessment plus transcriptome or focused off-target data | High-risk seed signature or passenger-strand activity |
| Delivery | Productive silencing in the intended delivery format and relevant cell type | Strong uptake but weak cytosolic activity |
| Developability | Defined purity, identity, stability, aggregation/particle attributes, and repeatable preparation | Activity depends on an unstable or poorly characterized preparation |
| Safety margin | Separation between pharmacologic activity and cellular/innate immune toxicity | Cytokine or viability effects near the efficacious range |
Later-stage packages should connect pharmacokinetics, tissue distribution, intact guide exposure, pharmacodynamic knockdown, and safety findings. Oligonucleotide DMPK and toxicity assessment are not end-stage add-ons: early assays can reveal chemistry- or delivery-dependent accumulation, metabolites, organ liabilities, and dose limitations before a program commits to a single lead.
Published Data
Case 1: Chirality-Dependent siRNA Modifications for Nuclease Protection
This 2020 original research paper (published in Nucleic Acids Research) explores the precise chemical design of siRNA therapeutics, specifically focusing on the synthesis and biological properties of introducing chiral modifications—5'-(R)- and 5'-(S)-C-methyl-guanosine—into siRNA sequences.
- The Goal: To introduce a methyl group at the 5'-carbon of terminal siRNA nucleotides and strictly control its stereochemical configuration (chirality). The objective is to create highly specific, position-dependent structural roadblocks (steric hindrance) against exonuclease degradation—an advanced chemical modification strategy that had previously been underexplored in RNAi drug design.
-
The Finding: The researchers discovered a profound, chirality-dependent protection mechanism based on exactly where the modification is placed:
- The (R)-isomer: When positioned at the 5' end of the siRNA, it significantly blocks 5'-exonucleases from degrading the strand because the enzyme cannot bypass the specific spatial orientation of the (R)-C-methyl group.
-
The (S)-isomer: Conversely, when positioned at the 3' end, it provides robust protection against 3'-exonucleases.
Both modifications successfully maintained the gene-silencing potency of the siRNA. Crucially, the corresponding 5'-triphosphate metabolites were not recognized as substrates by mitochondrial DNA polymerases, effectively minimizing the risk of mitochondrial toxicity.
- Practical Takeaway: This study proves the powerful viability of a "one modification type, two stereoisomers, dual-end protection" strategy. By selectively matching the (R)-isomer to the 5' end and the (S)-isomer to the 3' end, drug developers can achieve precise, bidirectional resistance to nuclease degradation while perfectly preserving the siRNA's target suppression activity and metabolic safety.
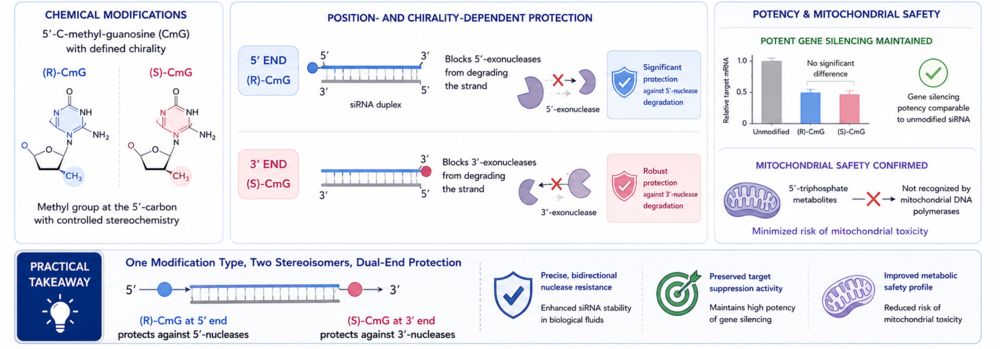 Figure 2. Stereochemical effects of chiral modifications on siRNA stability.
Figure 2. Stereochemical effects of chiral modifications on siRNA stability.
Frequently Asked Questions
Q: How many siRNA sequences should be screened for one therapeutic target?
A: There is no universal number. A practical first panel often includes multiple diverse sites across the relevant transcript rather than closely related candidates from one region. The appropriate panel size depends on isoforms, allele selectivity, transcript homology, chemistry, delivery format, assay throughput, and the cost of downstream profiling.
Q: Should chemical modifications be added before the first potency screen?
A: A two-stage approach is often useful. Initial screening can identify active sequence regions, but promising candidates should be retested early with the intended therapeutic modification pattern. Modification position can change Ago loading, potency, stability, immune recognition, and off-target activity, so unmodified results should not be treated as final rankings.
Q: What is the most important siRNA off-target mechanism?
A: Both near-perfect complementarity and miRNA-like seed activity matter. Ago2 can cleave highly complementary unintended transcripts, whereas guide or passenger seed regions can repress many partially matched transcripts. Risk assessment should therefore combine whole-transcript alignment, seed-match analysis, expression context, passenger-strand evaluation, and experimental RNA profiling.
Q: Does high cellular uptake guarantee effective siRNA delivery?
A: No. Total uptake may represent material trapped on the cell surface or retained in endosomes. Therapeutic activity requires cytosolic release, productive Ago2 loading, and access to the target transcript. Delivery studies should pair uptake and localization measurements with concentration-dependent mRNA and protein knockdown.
Q: Can the same siRNA be used in human cells and animal efficacy studies?
A: Only when the target sequence and relevant biology are sufficiently conserved. A human-specific siRNA may not bind the animal ortholog, while a surrogate sequence may differ in potency or off-target profile. Programs should define whether the animal study tests the clinical candidate, a species-matched surrogate, or only the delivery platform.
Q: What evidence is needed before nominating an siRNA lead?
A: A lead should show reproducible knockdown of mRNA and protein, a disease-relevant functional effect, activity in the intended chemistry and delivery format, acceptable transcriptome selectivity, a workable safety margin, and analytical quality suitable for repeat studies. The depth of evidence should match the intended next development stage.
Overview of What Creative Biolabs Can Provide
Creative Biolabs supports siRNA therapeutic development across candidate generation, experimental screening, delivery optimization, and preclinical evaluation. The appropriate development strategy depends on the primary challenge of the program, whether it involves sequence potency, chemical modification, target-cell delivery, pharmacokinetic behavior, or safety. The related capabilities outlined below can be integrated according to the project stage and the evidence required for lead selection or further translational development.
| Research Need | Related Creative Biolabs Support | How It Connects to the Current Resource Topic |
|---|---|---|
| Generate sequence-diverse candidates | Custom siRNA Synthesis | Produces traceable duplex panels and chemistry variants for comparative testing. |
| Rank functional candidates | siRNA In Vitro Screening Service | Measures concentration-dependent knockdown and supports lead narrowing in relevant cells. |
| Select and optimize a carrier | Delivery Method Development Service for RNAi | Aligns formulation or conjugate choice with target tissue, route, dose, and duration. |
| Develop hepatocyte-targeted conjugates | N-Acetylgalactosamine (GalNAc) | Supports receptor-directed liver delivery strategies and conjugate optimization. |
| Develop particulate delivery | Custom Nanoparticles Service | Enables evaluation of particle protection, uptake, endosomal release, and functional silencing. |
| Characterize systemic disposition | Oligonucleotide Drug DMPK Service | Connects intact guide exposure, metabolites, biodistribution, and pharmacodynamic duration. |
| Assess safety liabilities | Oligonucleotide Drug Toxicity Assessment Service | Examines chemistry-, sequence-, and delivery-related tolerability risks. |
| Interpret on- and off-target expression | RNA Profiling Service | Supports transcriptome-level evaluation of mechanism, seed signatures, and pathway effects. |
Researchers can contact us today to discuss the target product profile, available sequence information, intended delivery route, and the evidence required for the next development decision.
References
- Angart P, Vocelle D, Chan C, et al. Design of siRNA therapeutics from the molecular scale. Pharmaceuticals, 2013, 6(4): 440-468. https://doi.org/10.3390/ph6040440 Distributed under Open Access license CC BY 4.0, with modification.
- Mikami A, Erande N, Matsuda S, et al. Synthesis, chirality-dependent conformational and biological properties of siRNAs containing 5'-(R)-and 5'-(S)-C-methyl-guanosine. Nucleic Acids Research, 2020, 48(18): 10101-10124. 10.1093/nar/gkaa750