Glycosylation Disorder Solutions
Congenital disorders of glycosylation (CDG) represent one of the most challenging groups of inherited metabolic diseases, affecting protein and lipid glycosylation processes throughout the human body. As a leading biological specialist at Creative Biolabs with over two decades of experience in glycoprotein research, we are pleased to introduce the solutions for glycosylation disorders. At Creative Biolabs, we provide a robust, research-use-only platform tailored for glycosylation analysis, glycoprotein biomarker discovery, and disease profiling.
What Are Glycosylation Disorders?
Glycosylation disorders refer to a broad class of inherited or acquired diseases in which errors occur in the synthesis, processing, or transport of glycans. The most well-known group is CDG, a group of rare, multisystemic genetic diseases resulting from mutations in genes that control N-glycosylation, O-glycosylation, or glycan transport. The most commonly reported CDG subtypes in descending order include:
-
PMM2-CDG (phosphomannomutase-2 deficiency)
-
FKTN-CDG (fukutin-related disorders)
-
EXT1/EXT2-CDG (multiple hereditary exostoses)
-
ALG6-CDG (α-1,3-glucosyltransferase deficiency)
-
PIGA-CDG (phosphatidylinositol glycan anchor biosynthesis defects)
PMM2-CDG, being the most common form, has well-characterized clinical features. Patients typically present with severe multisystem involvement, including neurological dysfunction, coagulation abnormalities, and endocrine disturbances. Antithrombin deficiency is particularly common, occurring in 83.3% of patients, with 62.5% having activity levels below 50% of normal. This coagulopathy contributes to both bleeding tendencies (16% of patients) and thrombotic episodes (10% of patients).
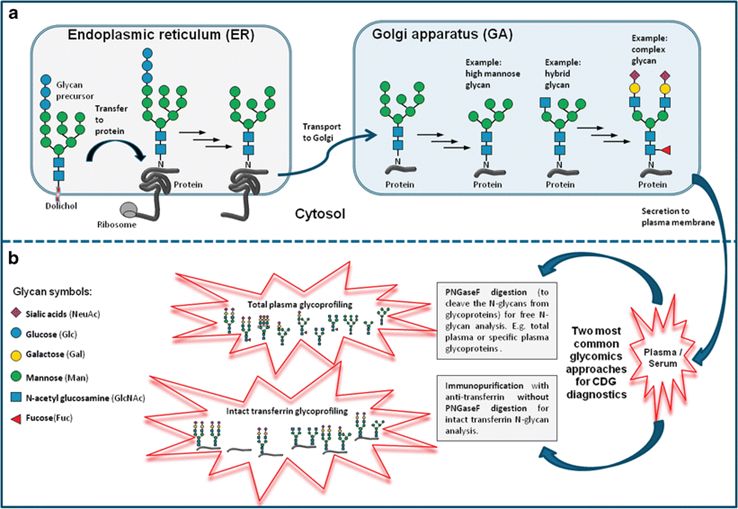 Fig.1 Overview of N-glycosylation, N-glycan types, and MS-based CDG analysis.1
Fig.1 Overview of N-glycosylation, N-glycan types, and MS-based CDG analysis.1
Classification and Types of CDG
CDGs are broadly classified into four main categories based on the affected glycosylation pathway:
|
Category
|
Pathway Affected
|
Key Features
|
Common Examples
|
|
Type I
|
N-linked glycosylation assembly
|
Transferrin type I pattern
|
PMM2-CDG, ALG6-CDG
|
|
Type II
|
N-linked glycosylation processing
|
Transferrin type II pattern
|
MGAT2-CDG, MAN1B1-CDG
|
|
Type III
|
Combined N- and O-linked glycosylation
|
Mixed glycosylation defects
|
COG-CDG subtypes
|
|
Type IV
|
Lipid and GPI anchor biosynthesis
|
Membrane protein defects
|
PIGA-CDG, PIGL-CDG
|
Creative Biolabs: Advanced Glycosylation Analysis Solutions
As a leading biotechnology company, Creative Biolabs offers comprehensive glycosylation analysis services that support research in CDG. Our advanced platforms provide critical insights into glycoprotein structure and function, enabling better understanding of disease mechanisms and therapeutic targets. We offer a suite of services, including but not limited to:
Our quantitative glycoproteomics platform allows precise detection and profiling of aberrant glycosylation in serum and cell-based samples. This platform is suitable for:
-
Identification of abnormally glycosylated serum proteins such as transferrin and haptoglobin
-
Glycoform profiling of fucosylation and sialylation changes
-
Monitoring of disease progression and therapeutic interventions in CDG models
-
Enzymatic glycan release using PNGase F
-
HILIC enrichment for glycopeptide isolation
-
High-resolution mass spectrometry for structural characterization
-
Site-specific mapping of serine/threonine modifications
-
Chemical derivatization for enhanced detection
-
Quantitative glycoform analysis
-
Comprehensive identification of modification sites
-
Quantitative assessment of site occupancy
-
Microheterogeneity analysis
Specialized Services:
Why Choose Creative Biolabs?
✔ End-to-End Glyco-Analytical Pipeline
We offer a modular service architecture:
-
Sample Preparation
-
Glycoprotein Enrichment
-
Glycan Structural Analysis
-
Bioinformatics Interpretation
-
Custom Reporting
✔ Disease-Focused Expertise
Our team has deep domain knowledge in:
-
Rare metabolic syndromes
-
Pediatric neurological disorders
-
Oncoglycomics (tumor-associated glycosylation)
-
Immunoglycomics (IgA nephropathy, autoimmune CDGs)
✔ Multi-Platform Integration
We integrate lectin microarray, MS, and glycan-release workflows under one umbrella, enabling cross-validation and higher analytical confidence. Our hybrid strategies overcome the limitations of single-method assays.
✔ Collaborative Approach
Our experts work closely with clients to define goals, select optimal strategies, and provide consultative guidance at every stage—from experimental design to data interpretation and manuscript preparation.
Understanding CDG congenital disorders of glycosylation demands robust analytical systems capable of resolving minute glycan variations. Creative Biolabs delivers a versatile platform for glycosylation biomarker screening, signature library building, and glycoproteomic profiling, advancing research in rare disease biology. Our non-clinical services empower scientists to pinpoint disease-specific glycan structures, differentiate CDG subtypes, and build the foundation for future glyco-therapeutic innovations. To learn more or initiate a custom glycosylation profiling project, contact our experts at Creative Biolabs today.
FAQs
Q: How can Creative Biolabs help identify specific glycosylation defects in CDG?
A: At Creative Biolabs, we offer a dedicated glycoproteomic analysis platform that enables high-resolution detection of disease-specific glycan abnormalities associated with CDG. Our integrated approach combines lectin microarray screening, quantitative glycoprotein profiling, and mass spectrometry-based glycan structural elucidation. For example, in PMM2-CDG, we assess transferrin isoforms using LC-MS/MS to detect increased disialotransferrin and absent tetrasialotransferrin, which are hallmark diagnostic features. Additionally, our platform can identify high-mannose glycans, core fucose deficiency, or hypersialylation patterns in serum glycoproteins or cell lysates. These insights help clarify the underlying enzymatic defects and provide a foundation for biomarker development.
Q: What types of biological samples do you accept for glycosylation disorder analysis?
A: We accept a range of research-grade samples, depending on the study objectives. Common sample types include:
-
Human serum or plasma – Ideal for transferrin glycoform analysis and global glycoprotein profiling.
-
Cell lysates or tissue homogenates – Suitable for mechanistic studies or cell model validation.
-
Cultured cells (fibroblasts, HEK293, etc.) – For experimental glycoengineering.
Each sample type is processed under optimized conditions to preserve native glycosylation patterns. We provide detailed collection and shipping guidelines to ensure data quality and reproducibility.
Q: Are your glycosylation disorder analysis services suitable for animal models or engineered cell systems?
A: Yes. We routinely support glycosylation analysis in murine models, CHO cells, and gene-edited iPSC-derived cells mimicking CDG conditions. Our workflows are fully adaptable to species-specific glycan structures and protein sequences.
For transgenic or knock-in models, we offer parallel analysis in serum and organ lysates to study tissue-specific glycosylation defects.
Reference
-
Abu Bakar, Nurulamin, Dirk J. Lefeber, and Monique van Scherpenzeel. "Clinical glycomics for the diagnosis of congenital disorders of glycosylation." Journal of inherited metabolic disease 41 (2018): 499-513. Distributed under Open Access license CC BY 4.0, without modification. https://doi.org/10.1007/s10545-018-0144-9
Resources
For Research Use Only.
