Peptide-Modified Liposomes:
Screening and Conjugation Strategies to Enhance Cellular Uptake
Overcome the translational hurdles of ligand selection and surface engineering to achieve true receptor-mediated internalization for targeted nanotherapeutics.
Common Bottlenecks in Peptide-Modified Liposome Development
Why Peptide-Modified Liposome Projects Stall
- Many peptide ligands bind well in solution, but lose accessibility or receptor-triggering activity after surface conjugation.
- Screening and conjugation are often optimized separately, leading to constructs that show binding without uptake or modification without reproducibility.
- Surface presentation parameters, including ligand density, linker length, and conjugation site, can increase nonspecific adsorption and compromise targeting performance.
Who May Benefit Most
- Teams developing receptor-targeted liposomes for oncology, inflammatory disease, or extrahepatic nucleic acid delivery.
- Biotech groups validating receptor-mediated internalization and downstream intracellular trafficking.
- Programs with an established liposome formulation that require optimization of ligand selection and surface conjugation parameters.
Empowering Liposomes with Peptide Ligands
Liposomes are widely used delivery vehicles with tunable composition and good biocompatibility. However, accumulating at a target site does not necessarily translate into efficient cellular uptake. While passive targeting strategies facilitate liposomal accumulation in solid tumors or inflamed tissues, encapsulated payloads—particularly nucleic acids or highly potent small molecules—must physically cross the plasma membrane to exert their effects.
To bridge this gap, surface functionalization with active targeting ligands is essential. Peptides are attractive ligands because they are smaller than antibodies, easier to synthesize, and more amenable to surface engineering. This reduced size minimizes the risk of premature immune clearance and allows for a higher ligand density on the liposomal surface.
Despite these benefits, modifying liposomes with peptides requires careful optimization. Identifying a peptide is only the first step; the formulation must also be engineered to preserve the ligand's activity. This challenge is particularly relevant for teams that already have a workable liposome core but need to improve ligand selection, receptor-mediated uptake, and surface-display reproducibility before advancing the formulation. Partnering with experts through a Targeted Liposome Development Service can help navigate these complexities.
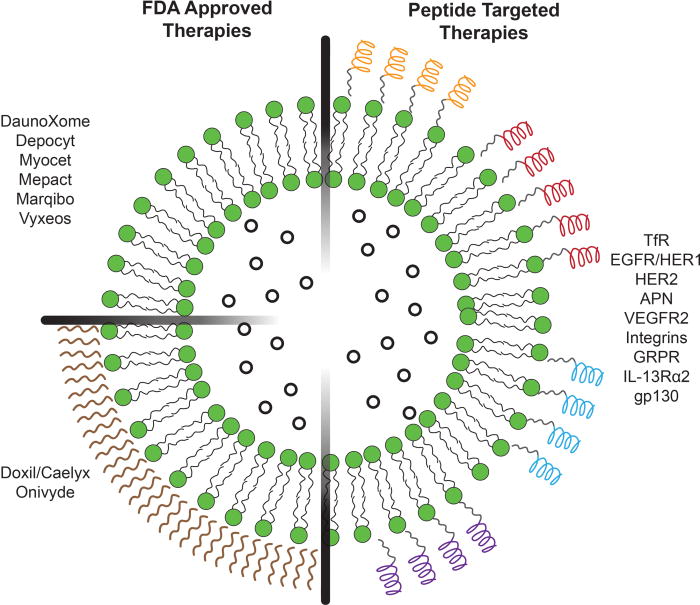
Peptide Screening: Prioritizing Internalization Over Mere Binding
A pervasive pitfall in targeted nanocarrier development is conflating receptor binding with cellular internalization. Traditional ligand screening platforms inherently bias the selection toward identifying sequences with the highest absolute binding affinity (Kd). However, strong binding on a two-dimensional axis does not invariably translate into efficient three-dimensional endocytosis.
Often, a peptide might bind tightly, anchoring the liposome to the cell surface, but fail to trigger the crucial intracellular signaling cascade required for membrane invagination. For therapeutic efficacy, the selected ligand must induce a conformational shift in the receptor that signals for active uptake—routing the liposome through appropriate endocytotic pathways.
To identify peptides with authentic uptake value, researchers must transition from static affinity assays to dynamic, functional in vitro internalization screening early in the discovery pipeline. Furthermore, the structural topology of the peptide must be rigidly assessed. Linear peptides are highly flexible and frequently lose their active conformation when tethered to the steric bulk of a liposome. In contrast, cyclic peptides—stabilized by internal disulfide bonds or synthetic bridges—provide superior resistance to proteolytic degradation and maintain a pre-organized architecture.
Common Screening Bottlenecks
-
1
The Binding Illusion
High affinity does not guarantee uptake. Ligands must trigger endocytotic signaling pathways.
-
2
Conformational Collapse
Peptides identified in solution often lose their active 3D structure once restricted by a conjugation linker.
-
3
Proteolytic Degradation
Failure to screen for serum stability leads to rapid enzymatic cleavage of the targeting sequence in vivo.
A Practical Workflow for Peptide-Modified Liposome Optimization
Systematically transition from free-peptide discovery to a fully validated targeted liposome using this integrated framework.
Shortlist Candidates
Select peptide candidates based on clear receptor relevance and a known mechanism of internalization rather than binding affinity alone.
Evaluate Free-Peptide Function
Assess the free-peptide's binding and active uptake simultaneously in both target-positive and target-low cell lines.
Select Conjugation Site
Engineer a conjugation site (e.g., terminal cysteine) that preserves the receptor-binding motif and ensures outward orientation.
Compare Linkers & Density
Synthesize liposomes with varying linker designs and surface densities to find the optimal balance between targeting avidity and stability.
Confirm Specificity
Validate the targeting specificity through rigorous competition assays or receptor-blocking in vitro studies.
Reassess Formulations
Continuously reassess particle size, zeta potential, stability, and batch-to-batch uptake consistency post-conjugation.
Conjugation Strategies: Geometry, Density, and Linker Design
Integrated Development Approach
In practice, peptide screening should not be separated from conjugation strategy design. A ligand that performs well as a free peptide may lose receptor accessibility, orientation, or uptake-driving activity once anchored onto a liposomal surface. For this reason, candidate prioritization is most effective when peptide internalization data, conjugation site selection, spacer design, and surface-density optimization are evaluated as one integrated workflow.
Implementing suboptimal chemistry can compromise the biological activity of the peptide, disrupt the structural integrity of the lipid bilayer, or trigger premature payload leakage. Establishing optimal surface display parameters requires exacting control over the chemical conjugation site, the spacer length, and the overall ligand density. Overcoming these chemistry-related hurdles is a core focus of Peptide-Modified Liposome Development.
The structural geometry of the attachment is paramount. Random conjugation—such as utilizing EDC/NHS chemistry to couple primary amines—often results in a heterogeneous liposome population where the receptor-recognizing domain of the peptide may be obscured or physically deformed. Directed coupling methods, such as utilizing Maleimide-Thiol coupling via an engineered terminal cysteine, can help improve the outward orientation of the peptide molecules.
| Conjugation Chemistry | Target Functional Group | Primary Advantage | Considerations |
|---|---|---|---|
| Maleimide-Thiol | Sulfhydryl (-SH) / Cysteine | Highly site-specific; excellent orientation control. | Requires reduction of disulfides; prone to retro-Michael reactions in vivo if not optimized. |
| Click Chemistry (DBCO/Azide) | Azide / Alkyne | Bioorthogonal; rapid kinetics; extremely stable linkage. | Requires unnatural amino acid incorporation into the peptide sequence. |
| EDC/NHS Coupling | Primary Amines (-NH2) | Simple, widely accessible reagents; no peptide modification needed. | Random orientation; high risk of destroying the peptide's active binding site. |
Beyond chemistry, the physical dimensions of the linker dictate accessibility. The spacer is often evaluated at lengths like PEG2000 to extend the peptide beyond the liposome's fixed hydration layer, effectively presenting the ligand to the cell surface. Conversely, an excessively long spacer increases the hydrodynamic radius and can cause peptide folding or entanglement.
Finally, ligand density must be rigorously optimized. While increasing the peptide count theoretically enhances binding avidity, an over-densified surface dramatically alters the physicochemical profile of the liposome. High peptide densities often increase surface hydrophobicity or alter the Zeta potential, subsequently triggering rapid clearance by the MPS, which undermines targeting efficacy.
Validating Receptor-Mediated Internalization
Formulating the construct is only half the battle. Rigorous, multiparametric validation is mandatory to ascertain that any observed enhancement in cellular uptake is genuinely driven by receptor-specific endocytosis, rather than generalized electrostatic interactions.
Competitive Assays
Commonly used to prove specificity in vitro, cells are pre-incubated with an overwhelming excess of free peptide ligand or specific receptor-blocking antibodies. A subsequent reduction in the uptake confirms their reliance on the specific receptor pathway.
Endosomal Escape Tracking
Evaluating the intracellular fate is crucial. Confocal laser scanning microscopy (CLSM) utilizing organelle-specific dyes determines if the payload successfully escapes the endosomal compartment before lysosomal degradation neutralizes the therapeutic agent.
Physicochemical Profiling
Post-conjugation characterization ensures the baseline integrity remains intact. DLS and Zeta potential analysis help confirm that the peptide conjugation has not induced critical aggregation or drastic charge shifts that would ruin biodistribution in vivo.
Frequently Asked Questions
Cyclic peptides are generally preferred for liposome targeting. While linear peptides are easier to synthesize, they are highly flexible and often lose their active, receptor-recognizing conformation once tethered to the liposome surface. Cyclic peptides, stabilized by internal bonds, maintain a pre-organized 3D structure and exhibit superior resistance to proteolytic degradation in serum, leading to more consistent targeting in vivo.
There is no universal "optimal" density; it must be empirically determined for each peptide-receptor pair. While increasing ligand density can enhance multivalent binding avidity, over-densifying the liposome surface often alters the zeta potential and increases hydrophobicity. This can trigger rapid clearance by the mononuclear phagocyte system (MPS) before the liposome reaches its target, defeating the purpose of the modification.
Yes, significantly. If the conjugated peptides introduce excessive positive charge or highly hydrophobic domains to the liposome surface, the construct may bind nonspecifically to negatively charged cell membranes or serum proteins. This highlights the importance of evaluating spacer length, conjugation site, and density as part of an integrated workflow, rather than simply maximizing peptide attachment.
Relying on standard fluorescence microscopy or flow cytometry alone can be misleading, as they may only detect surface adherence. To confirm true internalization, researchers should employ competitive assays (pre-blocking the target receptors), utilize pH-sensitive fluorophores that only emit signal within acidic endosomes, or perform confocal imaging with specific intracellular compartment markers.
Site-specific chemistries are strongly preferred. Methods such as maleimide-thiol coupling (targeting a terminal cysteine residue) or bioorthogonal click chemistry help ensure a uniform, outward orientation of the peptide ligand. This prevents the active receptor-binding domain from being obscured or structurally deformed, a common issue encountered when using random amine coupling (e.g., EDC/NHS chemistry).
References
- Aronson, Matthew R., Scott H. Medina, and Michael J. Mitchell. "Peptide functionalized liposomes for receptor targeted cancer therapy." APL bioengineering 5.1 (2021). https://doi.org/10.1063/5.0029860
- Under Open Access license CC BY 4.0, without modification.
Online Inquiry
This site is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.
